- History Home
- People, Leadership & Service
- A Legacy of Excellence
- History & Impact
- Meetings Through the Years
- Resources
Memoir - Richard E. Marsh (1922 - 2017)Memoir | Publications | Curriculum Vitae | Videos | Slides | Articles | Obituary
My Crystallographic History Richard E. Marsh 2013
Mooney had, a few years earlier, been accepted into graduate school at Caltech (my undergraduate school). But when Mooney arrived at Caltech, both sides were surprised: Caltech, because Mooney was a female (the "R" turned out to be "Rose"); and Mooney, because she did not know that Caltech, at that time, did not accept female students. What to do? My understanding is that Linus Pauling gave her a temporary appointment as research associate and helped arrange for her to transfer to the University of Chicago as a student of Will Zachariasen. She received her PhD there. After teaching at Newcomb she moved to MIT, where she later married the noted physicist John Slater.
Rose Mooney's office-laboratory-classroom contained a single x-ray tube and a Laue film holder — period. But she was a fine teacher, and I greatly enjoyed her crystallography class. As I recall, at that time Tulane did not offer a PhD program in chemistry, and after one year I was accepted at UCLA. Helen and I were married on August 11, 1947; as a wedding present I took her on a honeymoon trip of two thousand miles to the west coast, permanently displacing her from her family, all her friends, and the only city she had ever lived in. She seems to have forgiven me.
After my first year at UCLA I chose as my advisor Jim McCullough, who worked with organic selenium compounds. (He liked to tell stories about his evening trips home from his lab, and how all the other passengers moved to the far end of the streetcar when he climbed aboard.) I stayed far away from synthesis, but eagerly helped with McCullough's crystallographic research. One of his recent graduate students had been Gabrielle Hamburger (later, Donnay), and it was somehow comforting to have Gabrielle Hamburger, Martin Buerger, and C. W. Bunn so active in crystallography at that time.
At first our Fourier and structure calculations were carried out with pencil and paper, and with slide rules and logarithms. But soon we obtained a set of Patterson-Tunnell(1) strips, which were similar to the world-renowned Beevers-Lipson strips, for carrying out Fourier summations. Mechanical calculators were also becoming available. Some would even do division, which caused large clanks as the carriage bumped up and down in moving from column to column. Servicemen from Friden, Marchant, or Monroe suppliers were in frequent demand. I received my PhD at UCLA in 1950, the year after the ACA was founded. (My thesis was described as "sophomoric" by one of the committee members.) I then accepted a post-doctoral research appointment at Caltech, and held such an un-tenured position until I became emeritus in 1990.
The post-war years were tremendously exciting for diffraction crystallographers. The influx of students under the GI bill and the availability of funding from such agencies as the Office of Naval Research and the National Foundation for Infantile Paralysis led to rapid advances in diffraction equipment, structure determination and refinement methods, and particularly in computing capabilities. As early as 1947 Ray Pepinsky, at Penn State, developed XRAC (2) — a two-dimensional array of sine-wave generators where the amplitudes and phases could be adjusted and combined to present, on an oscilloscope, a two-dimensional Fourier map. It was in constant demand from crystallographers everywhere. In 1950 SWAC was installed and maintained by Ken Trueblood and Bob Sparks at UCLA. It contained over 2000 vacuum tubes, and was programmed for least-squares refinement (3). As I recall, it would play "The Star-Spangled Banner" correctly when all the tubes were working. The age of computers had begun.
Crystallography was particularly exciting at Caltech, because of Linus Pauling. The Nature of the Chemical Bond had been published in 1948, and he and Bob Corey were preparing to publish their descriptions of the alpha-helix and the beta-sheet. Many graduate students, postdoctoral fellows, and visitors from other institutions all over the world were at Caltech, working on structural problems of all sorts. (In 1956 Jack Dunitz, Leslie Orgel and Alex Rich published a joint paper (4) from Caltech on the structure of the new aromatic compound ferrocene.) A special occasion, in 1953, was a conference in Pasadena on protein structures with attendees from around the world. I shall not forget one occasion. A young scientist whose name I don't recall gave a dry and, perhaps, confusing presentation on methods of estimating the scaling factor for diffraction intensities from globular proteins. At one point he showed a slide of the relationship between I(q) — the measured intensity — and the diffraction spacing q. There was a question from the audience. It was W. L. Bragg: "Doctor, just what is your I(q)?"There was a long silence.
My first published paper from Caltech was with Pauling, on the structure of chlorine hydrate (5) — one member of a number of crystalline gas hydrates stable somewhat above 0 °C. Pauling, through his studies on intermetallic compounds, had developed a truly amazing insight into symmetric polyhedra, and he found that a polyhedron with twelve pentagonal faces and two hexagonal faces — a tetrakaidecahedron — and with equal edges of about 2.7 Å (a typical O-H···O bond) would fit into a cubic lattice so as to create a cage of water molecules able to accommodate the Cl2 molecule. This cage approximated an oblate spheroid with a diameter at the equator large enough to hold a Cl2molecule but with a polar diameter slightly too small. In order to approximate the disordered arrangement of Cl atoms — bonded together and with populations decreasing with latitude — we resorted to Bessel functions to calculate the diffraction intensities. The R for comparing calculated and observed intensities was 0.32. While this value seems high, it was based on intensity estimates from several 30° oscillation photographs (prepared in a cold room at 4°C) which consisted of powder lines superimposed with many single-crystal reflections; estimating intensities was difficult and imprecise. Other models devised by us and other workers gave notably worse agreements.
As I think back to the time that I spent in that cold room with frozen, fumbling fingers I remember Lindo Patterson, then at McGill University in Montreal, boasting of the low-temperature experiments he carried out during the winter using diffraction equipment mounted immediately outside his office window.
At that time I also worked on the structures of a number of intermetallic compounds, mostly with David Shoemaker (before he moved to MIT and married Clara Brink). They included NaPb (6), with its fascinating Pb4 tetrahedra; MgB2(7); Ag5Zn8 (8); and NaZn13 (9). The stoichiometry of such compounds interested quite a number of workers in the field who thought that the Fibonacci series might somehow be involved.
In 1952 Pauling saw a paper reporting the structure of the beta (monoclinic) form of selenium, Se8. He immediately knew it must be wrong. Rather than being an 8-membered ring as in the alpha (also monoclinic) form, one atom was displaced by about 1.1 Å so as to form a broken chain. It turned out that the problem could be corrected if the ycoordinate of that atom were changed from y to 0.5 - y. Since the structure determination was based on zero-level Weissenberg photographs (as was usual at that time), the only data that would be affected were the hk0 reflections with h odd. The original author had reported problems in interpreting the hk0 data, and the R(F) for them was 0.31. Measured intensity values for these reflections were not included in the paper, but a reproduction of the hk0Weissenberg photograph was; we were able to estimate intensities directly from it and obtained an R of 0.18 for our revised structure (10). This experience no doubt influenced my later interests in incorrect structures.
Dick Marsh and Linus Pauling at Pauling's 85th birthday celebration (Caltech, 1986).
A year or so later I became involved in a structural study on silk fibroin (11). This was a particularly interesting fibrous protein, since the worm's gland could be both stretched and rolled so as to achieve two-dimensional order, as expected for a beta-sheet structure. Weissenberg photographs indicated not only the repeat distance along the polypeptide chain but the spacings between the sheets. There were two such spacings — a small one to accommodate the glycyl and alanyl residues and a larger one to accommodate the remaining residues. I believe this model has survived the test of 60 years. A vivid remembrance of that study is the daily sessions of about one hour in Robert Corey's office, editing the one or two paragraphs of text that I had prepared the previous evening. Corey was a wise and gentle man, and he never once used the term "sophomoric".
At about the same time Joe Kraut, then a graduate student at Caltech, was spending happy hours at the local beaches collecting sea-gull feathers for his structural studies on feather-rachis keratin. As far as I know that rich, interesting diffraction pattern has not yet been satisfactorily explained.
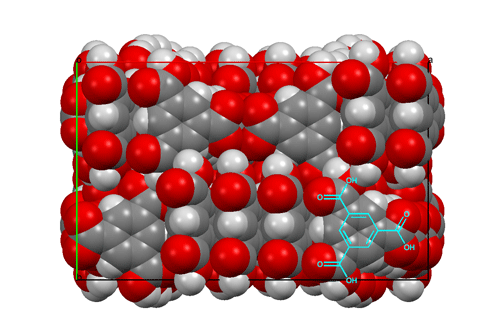
I should like to mention one other structure determination, from 15 years later; it was part of Dave Duchamp's PhD thesis. The compound was trimesic acid (12) — the symmetric tri-carboxylic acid derivative of benzene. Crystals were monoclinic, space group C2/c, with 48 molecules per cell (6 per asymmetric unit). This was a big computer chore for those times, but the structure was going to be interesting since an initial rotation photograph clearly showed the Fourier transform of a six-membered ring and h0ℓ Weissenberg photographs showed nearly exact mm symmetry. Complete data were collected, and the intensities of 11,563 independent reflections were estimated visually. As an aid to figuring out the structure, the Fourier transform of the molecule was generated from a cardboard mask placed in the path of a parallel light beam obtained from a pair of 12-inch lenses; comparing this optical transform with precession photographs suggested the orientations of the molecules in the a,c plane. The final least-squares refinement included 979 parameters and required about two hours on an IBM-7094 computer. All of this was carried out while Dave was developing the CRYM computing system of Fortran-based crystallographic programs. The resulting structure was based on six-membered rings of molecules hydrogen-bonded together to form pleated "chicken-wire" arrays; layers of three such arrays interpenetrated one another in a truly fantastic way. Maybe one can imagine Father Nature asking his wife, "How did you come up with that one, dear?" 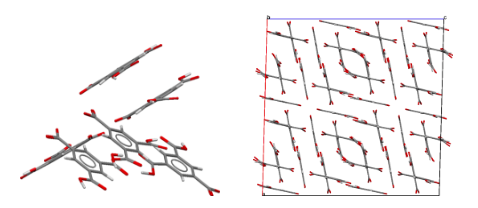 Views of the trimesic acid structure, D. J. Duchamp & R. E. Marsh (1969).
Packing diagram of trimesic acid, viewed down the c axis
For the past many years I have been concerned with the problem of incorrect structure determinations — almost invariably a mis-assignment of space group — appearing in the crystallographic literature. Small-molecule crystallography is, perhaps, unique in the amount of over-determination (in the ratio of observations to parameters) that it affords; there should be very little room for error. I somehow take such errors personally: they should not happen in MY field of study. Fortunately, in the past few years they have decreased in number, thanks to computer programs such as Checkcif and to slowly-successful pleas to journal editors to insure that authors make use of these programs. My recent surveys have suggested that the "wrong structure" disease may be getting close to extinction.
A couple of other highlights of my career:
In 1993 I served as president of the ACA. Fortunately, Bill Duax was the Chief Executive Officer, S. N. Rao the Treasurer, Marcia Colquhoun the Director of Administrative Services, and Judith Flippen-Anderson and Connie Rajnak the Newsletter Editors. The ACA is extremely fortunate to have them still in place.
For a few years I participated in the ACA summer course in crystallography, first at Pittsburgh with Bryan Craven and then at Athens, Georgia, with B. C. Wang. These courses seemed to me to be very valuable, for both the students and the instructors, and I am sure they remain very valuable today. A vivid memory from the 1998 session was watching Herb Hauptman, also an instructor in the course, in the middle of downtown Athens leading a group of about 50 local residents singing (in French) the Marseillaise on the day that France won the World Cup.
My 67-year tour through crystallography has been delightful and fulfilling beyond description. With very few exceptions, the people I have been associated with (far, far too numerous to mention) have been stimulating and delightful. I have truly been blessed. Thank you, R. C. L. Mooney.
Dick Marsh in 2012
REFERENCES (1) A. L. Patterson & G. Tunnell (1942). Amer. Min. 27, 655. (2) R. Pepinsky (1947). J. Appl. Phys. 18, 601 (3) R. A. Sparks, R. J. Prosen, F. H. Kruse & K. N. Trueblood (1956). Acta Cryst. 9, 350. (4) J. D. Dunitz, L. E. Orgel & A. Rich (1956). Acta Cryst. 9, 373. (5) L. Pauling & R. E. Marsh (1952). Proc. Natl. Acad. Sci. 38, 112. (6) R. E. Marsh & D. P. Shoemaker (1953). Acta Cryst. 6, 197. (7) M. E. Jones & R. E. Marsh (1954). J. Am. Chem. Soc. 76, 1434. (8) R. E. Marsh (1954). Acta Cryst.7, 379. (9) R. E. Marsh, F. J. Ewing, L. Pauling & D. P. Shoemaker (1952). Acta Cryst. 5, 637. (10) R. E. Marsh, L. Pauling & J. D. McCullough (1953). Acta Cryst. 6, 71. (11) R. E. Marsh, R. B. Corey & L. Pauling (1955). Biochim. et Biophys. Acta 16, 1. (12) D. J. Duchamp & R. E. Marsh (1969). Acta Cryst. B25, 5. |