- History Home
- People, Leadership & Service
- A Legacy of Excellence
- History & Impact
- Meetings Through the Years
- Resources
Memoir - Charles E. BuggMemoir | Publications | Curriculum Vitae | Videos | Slides | Articles | Obituary
ACA Living History2015
Download a printable PDF (10MB) version of this memoir here.
I was supposed to be an orthopedic surgeon, not a crystallographer. My father was a prominent orthopedic surgeon in Durham, North Carolina, where I was born and raised. As long as I can remember, I expected to follow in his footsteps. I was the second of five children. Except for brief stays in Fort Bragg, N.C. and Thomasville, Georgia, where my father was stationed during the war, my early childhood was spent in Durham. I was also destined to attend Duke University, where both of my parents, my grandfather and multiple other relatives attended college. My mother graduated Phi Beta Kappa from Duke in 1936. My father attended both undergraduate school and medical school at Duke. It was a done deal that I would attend Duke, especially since my parents made it very clear that they only intended to pay for me to go there. Fortunately, it proved to be a pretty good choice for me. My father had a private practice and was on the faculty in Orthopedics at Duke. My mother handled the finances for his practice. Orthopedic surgeons were fairly rare after the war, so my father eventually ended up treating many of the families of local farmers. He was well known throughout Durham and Orange counties. His number one recreation, which also became mine, was hunting and fishing, which were very productive activities in rural North Carolina back then. My father kept a hunting file close at hand in his office, and he would routinely get permission from patients to hunt on their land. It seemed that we had almost free range of the countryside for quail and dove hunting. We also had a rustic house on the North Carolina coast in an area that was totally undeveloped at the time, where the duck hunting was incredible. Fishing both in local lakes and on the coast was almost always productive as well. It was a wonderful time for me to grow up in the South. My father performed extensive charity work, which was fairly common for doctors in that era. He would frequently operate on weekends at the local charity hospital where conditions were incredibly primitive. I remember one weekend when he borrowed the hand drill from my junior tool set and autoclaved it to use in an operation because the hospital could not afford to purchase one. He sometimes would take me into the operating room with him, where he usually worked with no assistance, and he would explain the operations in detail. I remember one operation in particular. It was during the middle of the summer. Of course there was no air conditioning. A huge, obese woman had badly broken her hip, which needed to be nailed back together. In mask and gown I was suffocating, and the procedure was unusually messy due to the size of the patient. I recall that at the first time I began wondering if I really wanted to become an orthopedic surgeon. My mother was a strong influence in my life from the earliest times I can remember. She was very active in the Durham community, and she always seemed to have a secret game plan for my development. She did not micromanage my life, and she was always supportive, even when I did not deserve it. I initially attended Calvert School, now renamed Durham Academy, a private school where all of my close friends were enrolled. However, my mother was a strong advocate of public schools, and she served a number of years on the Durham School Board. Although I think my family could have afforded private school at the time, she moved me to Morehead School, a public elementary school, when I was in the fourth grade. This school was in a pretty rough neighborhood, and none of my friends or kids whom I had grown up with was enrolled there. It seemed that I was routinely roughed up every day after school, and I made it clear that I thought I really should return to Calvert. My mom's solution was to hire a retired, professional boxer to give me lessons in how to take care of myself. She sent me back into the jungle, where I finished elementary school. I actually ended up making some very good friends there, who had interesting backgrounds that I would have totally missed if I had stayed in private school. I never knew at the time exactly why my mother put me in situations like this, but it was clearly part of her plan. Maybe she wanted me to learn how to handle bullies later in life, and to get an up close view of another side of life. I had an unusual opportunity, again engineered by my mother, to get an early education in politics. At that time, before e-mail and electronic communications were available, the N.C. legislature relied on page boys to move paperwork back and forth between legislators in the State Senate and House. These page positions were typically assigned to elementary school kids, maybe because we were small and could easily wriggle between the rows of seats. Page positions were apparently highly valued, although any good reason for this escaped me at the time. Our local state senator, Claude Curry, was a family friend, and one of my cousins, William B. Umstead, was Governor of North Carolina while I was in the sixth grade, so it was probably pretty easy for me to be selected as a page in the N.C. Senate. The details of my absence were negotiated with Morehead School (by my mother, not by me); and every morning during the legislative session that year, I would either catch the Trailways bus to Raleigh or get a lift from Senator Curry and head to the legislative chambers. I would then have to catch up with the day's schoolwork in the evenings and weekends. I don't remember seeing much merit in this opportunity at the time, but it did teach me how to study on my own, and it ruled out politics as a future career. After elementary school, I attended public school at Carr Junior High, which was in the same general rough area of downtown Durham. I did not perform well in junior high, and it soon became clear that, if I really wanted to gain admission to Duke, even with whatever influence my parents might exercise, I would likely need to have a better education than I was getting in the Durham public schools. So I was shipped off to McCallie School, a military, preparatory school in Chattanooga, Tennessee, for my three years of high school. This is probably a main reason that I was eventually able to become a crystallographer, since McCallie had superb programs in math, chemistry and physics, with strict discipline that I badly needed at that stage of my life. I actually enjoyed my coursework for the first time, and I graduated in great physical shape having spent three years on the football and wrestling teams. During the summer vacations, I worked in construction as a manual laborer. I saved a lot of money during that period, since I lived at home and was usually too tired to do much at night. This became important later on when I married the love of my life at a fairly young age. I was admitted to Duke as a pre-med student in the summer of 1959. I enrolled in a 3-year program that would allow me to accelerate graduation by carrying a heavy course load and attending summer school. Since I needed multiple courses in chemistry for medical school, I selected chemistry as my major. I greatly enjoyed math at McCallie and figured it might require less study time, so I chose math as my minor. None of this was done with any thought about crystallography, which I didn't even know existed, but it turned out to be a pretty good curriculum for that eventual career. Better lucky than wise I guess. A real stroke of good luck was meeting Bebe Bradshaw on the first day of freshman orientation. She was introduced to me by a friend from McCallie, who was from Bebe's hometown of Winston Salem, N.C. Bebe and I became immediate friends, dated off and on, and finally fell madly in love. Her father was Chairman and founder of the Department of Surgery at Bowman Grey Medical School in Winston Salem. Since my father was on the faculty in surgery at Duke, he and Dr. Bradshaw knew each other, although Bebe and I had never met before Duke. Considering our similar backgrounds, it is not surprising that we were compatible and shared many similar views of the world. She was the most wonderful, intelligent and warm person I had ever met. We were pretty much inseparable during our last two years at Duke. She was and is my soul mate and has been a key support and driving force in all aspects of my life and career since those early years at Duke. We also enjoyed a wonderful social life at Duke, thanks to my grandfather, who had been a founding member of the Duke chapter of Kappa Alpha fraternity when he attended Duke. I am sure this was a serious, austere body when my grandfather was at Duke, but while I was there it was the Duke version of animal house. Their week-end activities were notorious, and to see my KA fraternity brothers on a Saturday night anybody would guess that we would all eventually end up homeless. In fact, most of them went on to stellar careers in various fields. One of my more famous classmates in the fraternity was Charlie Rose, who later became one of the most respected talk show hosts and commentators in the country. My claim to fame and popularity was that I installed and operated a still in the bathroom attached to my room in the fraternity house. Not surprising to me, this fraternity was banned from campus shortly after I graduated, but I had a number of close friends there who have remained an important part of my life over the years since Duke. Although the weekends were happily chaotic, Bebe and I made it through Duke in pretty good academic shape by confining the week nights to studying at her quiet dormitory on the Women's Campus (they were separated from the men back then, fortunately). We were determined to get married as soon as we graduated, despite her mother's vocal objections about our young age, and Bebe accelerated her curriculum in early childhood education, so that she would finish at the middle of her senior year. We were married at Christmas time, in 1962, at the age of 21, both with bachelor degrees from Duke and have since had a wonderful and exciting life together. I cannot imagine a life without her. If we had not had the good fortune of meeting at Duke Freshman orientation, I would probably still be doing manual labor in the construction industry. While at Duke, my goal of becoming an orthopedic surgeon was gradually replaced by my interest in science. I really liked chemistry and math but had a hard time enjoying anatomy and biology, which seemed to require brute force memorization. I probably would have found biology much more interesting in the current world of molecular biology; but back in 1959, the biology courses required for medical school were not all that exciting. I really was turned on by physical chemistry, thanks to a superb professor, Marcus Hobbs. During my senior year, I started having second thoughts about medical school. Surprisingly, I received strong encouragement from my father and from Bebe's father to pursue a career in science, and Bebe was supportive of following whatever path I found most exciting. Since physical chemistry was the discipline that I most enjoyed, I relied heavily on Professor Hobbs to steer me in the direction that he felt would be the best fit for me. He had good friends on the chemistry faculty at Rice University in Houston, Texas. After talking with them, he convinced me that their graduate program would be a great choice for me. The newly appointed President of Rice at that time was Kenneth Pitzer, a prominent physical chemist who had just arrived from Berkeley, and his influence was prominent in the Chemistry Department. One of his close colleagues from Berkeley, Robert Curl, who later shared the Nobel Prize in Chemistry for the discovery of buckeyballs, came with him and lectured in physical chemistry while I was there. Professor Hobbs arranged for me to be admitted to the Rice graduate program with what I considered a very attractive fellowship that would help support me and Bebe, once she could join me in Houston. I never thought about applying elsewhere and was excited about the opportunity to continue my studies at Rice. Following my graduation from Duke in the spring of 1962, I was fortunate to land a good summer job at the newly created N.C. Research Triangle Park. One of the early occupants of the park was Chemstrand Research Labs, a division of Monsanto Corporation. Chemstrand was the major synthetic polymer unit of Monsanto, with research programs focused primarily on the discovery and development of novel polymeric fibers. Synthetic polymer research was a hot area in the early 1960's (remember the movie "The Graduate"when Dustin Hoffman was advised that his main route to success was plastics?), and I felt very fortunate to get exposure to this area of chemistry. I spent the summer synthesizing and testing various derivatives of nylon. Each new batch that I synthesized was then spun into fibers. That process had considerable slack time, which freed me up to pursue other activities. I had not taken Russian, French or German while at Duke, and I knew that I would be required to pass exams in two of these languages as part of the PhD curriculum at Rice, so I studied French and German while babysitting the spinning test polymers that I had synthesized. It turned out to be an interesting summer which gave me a quick look at corporate research, taught me a little about polymers, positioned me to pass my future foreign language requirements, and added to my savings account. I arrived at Rice in the fall of 1962 and quickly decided on thesis research in physical chemistry. I was fortunate to be accepted as a student in the laboratory of Ronald Sass, a young, dynamic faculty member pursuing various research programs in crystallography. Dr. Sass was a really fortunate choice since he was at the stage in his career when he was especially enthusiastic about teaching. Dr. Sass also had a research grant from NASA to investigate the structures of organic semi- conducting materials. His grant provided a research fellowship for me, which freed me up to pursue fulltime research with no teaching or lab supervision responsibilities. Dr. Sass had obtained crystalline samples of a carbanion compound, pyridinium dicyanomethylide, with particular interest in the question of conformation around the central carbon atom. This small, light-atom compound would be a trivial crystallographic challenge in modern times, but in 1963, it was difficult to determine a crystal structure that was even that simple — especially for a wet-behind-the-ears graduate student. Automated diffractometers were not yet readily available, and most structures were still being determined using tedious film techniques. I quickly became an expert in Weissenberg photography and manually estimated the intensities of thousands of film spots by comparing each separately with diffraction spots produced on standardized film strips. Computing was also a major challenge at the time, but it was fortunate that the Department of Electrical Engineering at Rice had recently constructed a computer that was available at night and on weekends. This computer occupied a complete floor of the engineering school and was constantly breaking down. It probably had a tiny fraction of the power of a modern smartphone, but it beat calculating Fourier maps by hand. The small organic structure was solved by Patterson and packing analyses, and it was refined using many hours of computer time on the Rice computer [1-3]. That crystal structure was followed by determination of the structure of a related compound, potassium paranitrophenyl dicyanomethide, an analysis that was aided by the presence of the heavy potassium ion [1, 2]. These two crystallographic studies were the final subjects of my PhD thesis, which I completed in the spring of 1965. While I was at Rice, Bebe and I lived in an apartment complex that was only a few blocks off campus, and several of the other chemistry graduate students and spouses were there also. Bebe quickly obtained a job teaching fourth grade at one of the newer elementary schools on the edge of Houston. Houston was growing rapidly at that time, and she had 62 students in her first class with no assistant to help. A big portion of her kids spoke only Spanish, so she had to use other students to translate for her. Several of the other graduate students' wives in our apartment complex also taught elementary school, under equally taxing conditions, so we and our friends were really ready to blow it out on the weekends. I switched from running a still to making beer, since I had inherited all of the formulas and equipment for brewing from a graduating organic chemist. We had parties with homemade beer and popcorn about every weekend, which we could easily afford on our tight budgets. Three of us were avid duck hunters, and the Houston area was covered up with waterfowl in the rice fields within a few miles of the Rice campus, which was actually on the outskirts of Houston back in the early 1960's. We went duck and goose hunting together early each Wednesday morning during duck season and shared a large freezer locker that was always full of game. Bebe's mother had warned her that she would probably starve to death if she married me, so I was happy to prove her wrong. One of my fellow duck hunters dedicated his PhD thesis in organic chemistry to Morgan LeFleur, our duck-hunting guide, with a caption saying that he could not have made it through graduate school without Morgan's encouragement and guidance. I don't think his supervisor ever knew who Morgan was. At this stage, I did not know exactly what I wanted to do with the rest of my life. I seriously reconsidered going to medical school and discussed this with my father and my father-in-law. My father was a close friend of Philip Handler, the Chairman of Biochemistry at Duke, and he arranged for me to meet with Dr. Handler to discuss career options. This turned out to be a pivotal meeting for me. Dr. Handler, who was a prominent member of the U.S. Academy of Sciences, was charismatic, knowledgeable and persuasive in his view that crystallography was a wonderful opportunity for me in biology. He contended I would be wasting valuable time in my career by attending medical school. He told me of the forefront research underway in biological crystallography and urged me to join one of the major groups working in this field. He explained to me that the leading U.S. crystallography groups in biological crystallography were at MIT and Caltech, and he urged me to apply to one of those groups to continue my crystallography training. With help from Dr. Sass, a postdoctoral position was arranged at Caltech, in the laboratory of Dick Marsh and Bob Corey, and I joined them in the spring of 1965. I was really fortunate to end up at Caltech. The crystallography group was located in the Department of Biology, where exciting research in the new field of molecular biology seemed to be underway in every laboratory. Along with this rich biology environment, my crystallography training moved to an entirely new level under the supervision of Dick Marsh. Dick is a notorious stickler for high precision in all aspects of crystallographic structural studies, beginning with collection of accurate diffraction data and through the final writing of a proper manuscript describing the analysis and results. I like to think that much of his obsession with doing everything as perfectly as possible rubbed off on me during my time with him, and that I, in turn, have had some success in passing those principles on to my students and postdoctoral fellows. I do know that I immediately think of Dick, and suffer pangs of guilt, anytime I consider taking a short cut in experimental procedures or in properly analyzing and reporting crystallographic results. Under Dick's close scrutiny and encouragement, I redetermined the crystal structure of potassium paranitrophenyl dicyanomethide shortly after arriving at Caltech [4]. This proved to be a great lesson in how to do things the "Dick Marsh way" and allowed me to get quickly immersed in the data collection and computing facilities at Caltech. Computing facilities were especially state-of-the-art at Caltech, with a top line IBM mainframe and a large computing center support staff to help with even routine problems. I quickly immersed myself in mastering Fortran, which was the universal language of scientific computing in the 1960's. Back in those days, there was nothing equivalent to the modern apps that allow most computing functions to be performed automatically, and many of the computer programs required for routine crystallographic procedures were still being developed. If we wanted to do anything unusual, we had to write the software ourselves. The early exposure to computing at Caltech was a wonderful opportunity for me to acquire the software development experience that allowed me to eventually initiate a crystallography group of my own. After demonstrating that I was a believer in proper crystallographic procedures, I was ready to embark on my original goal of structural biology. Following the Watson-Crick discovery of the double helical structure of DNA, there was broad interest in better understanding the detailed atomic-level structures of nucleic acid components so that more precise models of nucleic acids could be developed. I was fortunate to obtain crystals of cytidylic acid, one of the four components of RNA, and the crystallographic analysis of that nucleotide became my first major project at Caltech [5]. This also began what eventually became a multi-year career in crystallographic studies of nucleic acid components and their analogs. After a year at Caltech, I was still not sure what I wanted to do with the rest of my life. I had enjoyed all aspects of my studies and research at Rice and Caltech, and during my summer in industry at the Research Triangle Park. The 1960's were a great time to be in science, and many career opportunities were available. I interviewed with several chemical companies and was especially excited by the broad research programs at DuPont. I ended up accepting a position with their polymer fiber division, at their research laboratories located in Kinston, North Carolina. This decision was probably driven largely by geography, since Kinston placed me and Bebe near our families and within an hour of the Bugg beach house on the coast. I also felt that my previous experience with polymeric fibers at Chemstrand gave me a little insight in what might be involved at DuPont. One immediate, unforeseen benefit of choosing DuPont arose shortly after I began working for the company. In 1965, the Vietnam War was heating up, and I was called up for the draft shortly after I arrived at DuPont. I passed the physical with flying colors, and was on the way to the Army when the Director of the Kinston laboratories intervened with the draft board. As it turned out, DuPont had military contracts for developing synthetic fibers to be used to produce novel fabrics for parachutes and other applications, and I was considered among the essential personnel for fulfilling these contracts. Although I felt a little guilty about not following some of my close friends to the war, I must admit that I was relieved when I was deferred from the draft. I thought it would probably be a temporary deferment, but I never heard from the Draft Board again. I did not actually work directly on parachutes, as far as I know, but I did become deeply immersed in several novel research programs at DuPont, all in the area of polymeric fibers. I had not been hired as a crystallographer, but much of my early research at DuPont involved characterizing new types of fibers and testing ways they might prove useful. The Kinston laboratories had X-ray diffraction equipment, which they mainly used for patent coverage based on characterization of fiber crystallinity, size of crystalline domains, and crystal orientation. They also had an IBM mainframe computer, mainly used for business applications, available to me with almost unlimited time on evenings and weekends. Although it was probably not what the laboratory director preferred me to be doing with my time, I ended up determining the crystalline structure of the unique polymer that my group was developing, using fiber diffraction data. I submitted a paper on this structure for management review in 1966 but have not yet obtained approval to submit the paper for publication. However, I did have two pairs of test pants tailored by the company and made from samples of this novel polyester (which produced a synthetic cashmere-type fabric). I probably wasn't supposed to do so, but I took them with me when I left DuPont and wore them for years when duck hunting in freezing weather. As far as I know, this polymer never made it to the market, even though I found it to be a wonderful new development. However, the project did give me a unique opportunity to learn about fiber diffraction analysis. I had many outstanding colleagues at DuPont, my salary was substantial, the company was generous in allowing me to pursue basic studies that were not in line with their major priorities of developing novel polymers, and the geography was perfect. Within six months, however, it was clear to me that a large company, even one as outstanding as DuPont, was not where I wanted to spend the rest of my life. I greatly missed the freedom and stimulation of academia. After several months at DuPont, I submitted an application to NIH for a postdoctoral fellowship to continue my studies of nucleic acid components. I was delighted when I was awarded the fellowship and was then faced with the decision of where to go for my continued postdoctoral research. I had been accepted in Alex Rich's laboratory at MIT, and I was also confident that I could arrange to return to Caltech. Bebe and I went to Boston to visit the Rich research group in mid-winter of 1966. There were two feet of snow on the ground at the time, and we were astounded at the price of housing in the Cambridge area. Plus, nobody showed up at the crystallography lab within hours of my pre-set appointment time. This was disappointing, even though I now understand that unusual hours were standard procedure for that group. I immediately contacted Dick Marsh who was happy to accept me back into his lab in sunny California. The second year at Caltech was one of the most productive periods of my life. I now had seen enough of the world to know that crystallography in academia was where I belonged, and I had enough scientific experience to jump feet first into meaningful research. I was especially fortunate to strike up a close friendship with Ulf Thewalt, a brilliant German postdoctoral fellow in the crystallography group. With Dick Marsh's constant enthusiasm and guidance, Ulf and I initiated several crystallographic studies of nucleic acid components that had not yet been analyzed, and ended up determining the structures of guanine, inosine and guanosine [6, 13, 14]. In modern times, these crystallographic studies would be routine using direct methods of phasing, but in 1967 it was a difficult job to solve the crystal structures of such light-atom compounds, especially those with chiral centers. However, it was a great way to learn more about crystallography, especially with the tremendous talent around for guidance in the Caltech group. Along with the continuous help from Dick Marsh, we also learned a tremendous amount about the latest diffractometer data-collection procedures under the tutelage of Sten Sampson. All of this turned out to be critically important in allowing me to later set up my own crystallography program in a new location. I started interviewing for university faculty positions early in 1968. My initial focus was on chemistry departments, since chemistry was most compatible with my background. Fortunately, many good universities were expanding into crystallography at that time, and there were a number of tenure track faculty positions available around the country. The 1960's and 1970's were a period of considerable growth in university science departments, and research funding was increasing at a much faster pace than in current times. It was a good time to be looking for a faculty position in crystallography, and I was in several advanced discussions after interviews with top chemistry departments when an unusual opportunity suddenly fell in my lap. The University of Alabama in Birmingham (UAB) received a large NIH grant to establish an interdisciplinary Institute of Dental Research in Birmingham, which was home to one of the top dental schools in the country. The focus of the grant was to recruit top scientists in basic science disciplines, in collaboration with the basic science departments in the medical and dental schools. In their grant application, they proposed to hire a crystallographer, and they had been awarded considerable funding that was available to support this position. When they suddenly received the grant, they really did not have a game plan in place for recruiting a crystallographer, but they knew that Caltech was home to Linus Pauling, a prominent crystallographer. The search committee called Caltech to speak with Dr. Pauling, but he was away at the time. Knowing that Dr. Corey worked closely with Dr. Pauling, they contacted him to ask his advice. They gave Dr. Corey a glowing, enthusiastic description of their vision for the future of research in Birmingham and their commitment to first class basic science, and they described the large, unrestricted source of funding they had available for recruiting faculty and equipping their laboratories. Dr. Corey came away from their conversation incredibly enthusiastic about this unusual opportunity. Since Dr. Corey knew I was from the south and probably would not immediately reject the idea of moving to Alabama, he contacted me and urged me to look at this opportunity. After talking with the search committee, I quickly arranged a visit to Birmingham and came away really excited about the prospect of joining the faculty there. Further reinforcement came from my father- in- law, who knew that Dr. John Kirklin, one of the most prominent cardiovascular surgeons in the country, had recently moved from Mayo to the position of Chairman of the Department of Surgery in Birmingham. My father- in- law strongly encouraged me to look seriously at this opportunity since he had heard many good things about the growth underway in Birmingham. After settling questions of laboratory space and funds for equipping a crystallography program, I accepted positions as Assistant Professor in the Department of Biochemistry, Investigator in the Institute of Dental Research, and Investigator in the Laboratory of Molecular Biology. Bebe and I moved to Birmingham in the spring of 1968, along with our first child (Jeannie), who had been born in Pasadena, and our weimaraner (Dixie). My long-range goal was to extend my earlier crystallographic structural studies to include a variety of nucleosides, nucleotides, oligonucleotides, purines, pyrimidines, and analogs of nucleic acid components. Although some crystal structures had been determined in this area, it was still virgin territory. Many of the detailed structural features that would be required for constructing meaningful models of nucleic acids, and understanding their multiple biological roles remained poorly defined. A number of analogs of nucleic acid components had been synthesized and shown to have important therapeutic value, but understanding the exact mechanisms by which these analogs alter the biological properties of nucleic acids would eventually require more detailed knowledge of their structural properties. Although my aim was to start immediately on crystallographic studies in Birmingham, I soon realized that it would be a number of months before my laboratories could be adequately remodeled and essential X-ray diffraction equipment installed, tested and ready for use. This down time proved to be an important opportunity for me to thoroughly analyze the crystal structures that had been determined and to better understand the important structural questions that needed to be addressed. Several other research groups were concentrating on analyzing the conformational features of nucleic acid components, but one area that seemed to be less understood was the interaction patterns among purines and pyrimidines. Hydrogen bonding patterns were generally understood, and this knowledge had been central in the Watson-Crick discovery of the DNA double helical structure. A striking feature of the crystal structures that we had determined at Caltech for guanine, inosine and guanosine was the intimate stacking of the planar purine rings, and it was generally appreciated at the time that stacking between adjacent base pairs in double helical DNA was an important stabilizing effect. However, little was known about the exact nature of the specific interactions that might be of importance in understanding these stabilizing effects or about how these interactions might be altered by incorporation of base analogs or intercalating compounds into DNA. With the help of Joe Thomas, a bright recent high school graduate who was on his way to study physics at the University of Michigan in the fall, I undertook a comprehensive analysis of the stacking patterns found in all the crystal structures that were then available for purine and pyrimidine derivatives. Our analysis was eventually combined with similar studies underway by M. (Sundar) Sundaralingam and his colleagues at Case Western Reserve University. The results of our analysis allowed us to better understand the interactions contributing to the stacking patterns found in nucleic acids and in crystal structures of purine and pyrimidine derivatives [9]. The analysis also served as a useful starting point for helping us select meaningful future crystallographic studies to better understand the specific forces governing base stacking interactions. Once I had a functioning crystallography lab, including a Picker single crystal diffractometer automated by a PDP-8i computer from Digital Equipment Corporation, I was ready to begin my crystallography career at UAB. I was extremely fortunate to be joined by my Caltech colleague Ulf Thewalt, who was eager to continue the fruitful crystallographic collaboration we had initiated in Pasadena. Our crystallography group undertook a variety of structural studies of purine and pyrimidine derivatives along with other molecules of biological interest [7, 10, 12, 15, 16, 20, 24, 32, 47]. We also initiated productive studies of calcium and phosphate complexes [27, 56], and compounds for better understanding the structural chemistry of phosphorous [8, 18], much to the joy of my colleagues in the dental field. Although my laboratory had been funded using the NIH grant that established the Institute of Dental Research, and I was physically located in Institute space, I was under no pressure to work on projects related to dentistry. In fact, the Institute had recruited a number of superb basic scientists in multiple disciplines who were pursuing forefront research projects in biochemistry, molecular biology and cell biology that had little to do with classical dental research. However, the general exposure that I had with dental research quickly led me to understand that relatively little was understood about the structural chemistry of calcium and phosphate or about the range of interactions that these ions had with proteins, carbohydrates, lipids, and other biological molecules. In addition to Ulf, I enjoyed the benefit of collaborating with another of my Caltech colleagues, Mani Subramanian, who joined my group shortly after Ulf departed for a new faculty position in Germany. The first few years in Birmingham were a wonderful journey in small-molecule crystallography with multiple structural studies focused on novel structures of nucleic acid components [26, 37, 58, 85, 92] purine and pyrimidine analogs [30, 35, 46, 49, 51, 52, 54, 55, 57, 61-63, 78, 84], calcium and phosphate complexes [17, 22, 23, 25, 28, 29, 33, 34, 36, 38, 40, 41, 44, 48, 50, 53, 59, 64, 69, 73, 74, 77, 80, 82, 97, 104], biological molecular interactions [19,42,43], and the structures of other small molecules of biological interest [11, 31, 39, 45, 60, 70, 71, 75, 79, 83, 86]. I think that these structural studies added significantly to the foundation for understanding the base stacking interactions of natural and modified purines and pyrimidines and the interactions that occur in biological systems between calcium and phosphate ions and various biological ligands. These crystallographic studies in Birmingham also expanded our understanding of how purine and pyrimidine analogs can perturb nucleic acid conformations and interactions. In addition to the continued friendship and collaboration with Ulf and Mani, our research during this period benefitted greatly from the hard work and creativity of several productive postdoctoral fellows and graduate students, including Bill Cook, Jerry Freeman, Rick Hearn, Helen Sternganz, Howard Einspahr, and Larry DeLucas. Howard Einspahr did a particularly beautiful job bringing together data from all of our calcium structures with other data from the Cambridge Structural Database to lay out a comprehensive picture of how calcium ions interact with various biological ligands [65, 66, 81, 89, 99]. In 1971, the UAB Cancer Center was designated one of the first Comprehensive Cancer Centers by the National Cancer Institute. The grant from NCI that funded our Comprehensive Cancer Center provided support to expand our crystallography program. I had a research grant from NCI to support our structural studies of purine and pyrimidine analogs, at the time we submitted our initial application to NCI to fund the Comprehensive Cancer Center, and I was designated to serve as the first Associate Director for Basic Sciences in the Center. We were also awarded funds to establish an X-Ray Crystallography Core Facility within the Cancer Center, which would be available to support collaborative structural studies with other Cancer Center members. This grant allowed us to expand our computing facilities within the crystallography group, develop a computer graphics facility, and hire additional postdoctoral fellows to be trained in structural biology. The grant also picked up a significant portion of my salary, which allowed me to devote more time to focus on crystallography and training of graduate students. Several of our colleagues in the Institute for Dental Research and the Comprehensive Cancer Center had research programs directed at isolating and characterizing important proteins, and they were constantly urging us to collaborate on protein structural studies. It was clear that we would have a number of exciting new directions we could go if we expanded our program into the rapidly developing field of protein crystallography. We had an especially productive collaboration at that time with John Montgomery, who was Director of the Organic Chemistry Division at nearby Southern Research Institute (SRI). John also held a joint appointment in our Cancer Center. He was well known in oncology, and several drugs developed in his laboratory were being used successfully in treating cancer patients. Among his numerous recognitions, John was a member of President Nixon's Cancer Advisory Board (Nixon had declared his War on Cancer which was the reason that funding had surged in cancer research), and John was excited about new protein targets that had been identified in cancer research. During his remarkable career, John had spent years trying to design compounds to inhibit enzyme targets in oncology without knowing the structures of the enzyme target sites, and he was constantly urging me to focus our crystallographic studies on some of the important protein targets in cancer. John had also introduced me to George Hitchings and Gertrude Elion at the Burroughs Wellcome Pharmaceutical Company, who later shared the Nobel Prize in Chemistry for Lifetime Achievements in Medicinal Chemistry, and they shared John's urgency to know more about the structures of the protein targets that they were pursuing. I entered into a consulting contract with the Burroughs Wellcome medicinal chemistry group to help with their small-molecule structural analyses, so I was well aware of interesting protein targets of interest to them. It became increasingly clear to me that we needed to expand our Birmingham program into protein crystallography if we were going to take full advantage of opportunities in our new Cancer Center. UAB had a policy of optional faculty sabbaticals every seven years, and I decided to use this opportunity to learn the essentials of protein crystallography. The University of Oxford had one of largest protein crystallography programs at the time, under the joint guidance of Dorothy Hodgkin and David Phillips. In addition, they initiated one of the first major university/industry joint programs in structure-based drug design, which was a collaboration between their protein crystallography group and Wellcome Pharmaceutical Company (the parent of Burroughs Wellcome in the U.S.) to design and develop compounds to modulate the activity of human hemoglobin. I applied to David Phillips to spend the 1974-1975 year with them and was delighted when they welcomed me. My colleagues at Burroughs Wellcome were also eager for me to go and offered me a grant to work on the structure of dihydrofolate reductase, a major drug design target within their medicinal chemistry program, during my sabbatical at Oxford. So, in the spring of 1974, Bebe packed up our three young children, and we took off for Oxford, along with 100 mgs of purified E. Coli dihydrofolate reductase from Burroughs Wellcome. The University provided us with housing close to campus, and my children were quickly enrolled in superb private schools, thanks to help from my Oxford colleagues. My lab at Oxford was located next door to Dorothy Hodgkin, who had received the 1964 Nobel Prize in Chemistry for the structures of penicillin and vitamin B12. She had transitioned to proteins and was then working on the structure of insulin. I was immediately at home and comfortable with Dorothy, who was incredibly warm and welcoming, and I felt that we shared a common bond in transitioning from small-molecule crystallography to protein crystallography. Dorothy also had several other colleagues in her group who were making similar transitions to proteins, so it was a great environment for me to begin this new career.
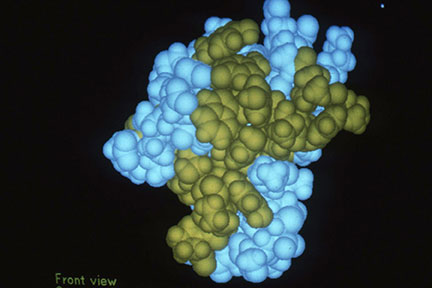
Our Cancer Center Grant at UAB was scheduled for renewal shortly after I returned from Oxford. Accordingly, I had a great opportunity to seek the extended funding that would be required to expand our program effectively into protein crystallography. I returned to Birmingham for a couple of weeks at mid-year to write the renewal proposal for the X-ray Crystallography Core Facility. We proposed in the grant renewal to hire another faculty member who had experience in macromolecular crystallography and to purchase the data collection equipment and computer modeling facilities that would be needed for protein structural studies. We were fortunate to hire Bud Suddath, who had recently completed several years with the Alex Rich group at MIT where he played a major role in determining the crystal structure of t-RNA. Bud had also been heavily involved in equipping the Rich laboratory for this type of crystallographic project, and he came to us with a superb background in crystallographic computing and in the fundamentals of macromolecular crystallography. Bud was also from the south and had done his undergraduate work at Georgia Tech, so he was immediately comfortable and enthusiastic about moving to Birmingham. We were successful with our NCI request for expanded funding to equip the laboratory for protein crystallography, and Bud and I went about the tasks of renovating additional space that the University generously made available for our expanded crystallography program, installing new equipment, and teaching the current graduate students and postdoctoral fellows what they would need to know for taking on new projects in protein crystallography. We had several different protein targets that we wanted to pursue at UAB, and we initiated multiple efforts to purify and crystallize these proteins for structural studies. The first protein structure that we actually completed was of a scorpion neurotoxin (Fig. 2) [72, 90, 93-95, 98, 100, 111, 131, 157]. We had entered into a productive collaboration with Dean Watt, from Creighton University, to study the very interesting proteins isolated from the venom of Arizona scorpions. Dean had devoted his career to isolating and characterizing these scorpion proteins which acted by binding to the sodium channels of nerve cells, and he was convinced that the three dimensional structures would be essential in understanding how the toxins modulate nerve impulses. Dean came to Birmingham and spent a year working with us on purification and crystallization of these proteins isolated from scorpion venom provided by a colleague at Arizona State University. The venom of the Arizona scorpion contains more than twenty different toxins that target the sodium channels of different animals, birds and insects, so they were very interesting probes for understanding variations in sodium channel structures among various species. The first toxin structure that we completed was an effort that benefitted greatly from the work of Bob Almassy, a brilliant postdoctoral fellow who joined us from Caltech, and from Juan Fontecilla-Camps, a graduate student who worked closely with Bob. The structural results provided a working hypothesis for how these proteins interact with membrane receptors and led to several additional studies designed to better understand the species specificity displayed by these proteins. Those early years after returning from Oxford also produced several other important protein structural results, including the crystal structures of ubiquitin (a protein that continues to be the focus of many biological studies due to the central role it plays in protein turnover) [76, 107, 119, 121], and calmodulin (a calcium-binding protein that regulates many biological processes, and continues to be of great interest in multiple areas of biological research) [87, 88, 102, 108, 125-127, 129]. In addition, preliminary crystallographic results were reported for pea lectin at low-resolution [96]; other scorpion toxins [103, 106]; sea anemone toxin [105]; human C-reactive protein [123, 138]; bacterial purine nucleoside phosphorylase [109]; human serum transferrin [68]; and porcine aldose reductase [148]. The protein crystallography projects in Birmingham during those early years of our program received valuable contributions from Bud Suddath, Bill Cook, Larry DeLucas, Howard Einspahr, Larry Gartland, Juan Fontecilla-Camps, and Bob Almassy. These crystallographers all went on to have remarkable careers in crystallography and molecular biology at UAB, other leading academic institutions and in industry. We also benefitted greatly from our multi-year collaboration with Dean Watt who added an essential biochemistry and protein purification capability to our group during those early years in protein crystallography.
Shortly after returning from my sabbatical in Oxford, John Montgomery and I began the process of selecting a suitable target for pursuing structure-based drug design guided by protein crystallography. This had actually been a major goal that strengthened our NCI renewal grant proposal for support of the Cancer Center, and there were many known protein targets in oncology that would be suitable for this approach. Since both John and I had considerable experience with purine and pyrimidine derivatives, including several that were useful chemotherapeutic agents in oncology, we focused our initial efforts on enzymes involved in purine and pyrimidine metabolism [115]. We soon settled on the human enzyme purine nucleoside phosphorylase (PNP) as a potentially ideal target for drug design. PNP had been demonstrated to be essential for normal immune responses since children born with defects in the gene for PNP lacked T-cell immunity. Inhibitors of PNP might prove useful clinically for treating T-cell mediated diseases, including a variety of autoimmune diseases, T-cell leukemias, and T-cell lymphomas. In addition, inhibition of PNP would block the biological synthesis of guanine from guanosine and could thus be used to inhibit the synthesis of uric acid, for treatment of gout. We knew that it would be a long and difficult road through the crystallographic studies, and through the eventual design, synthesis and development of inhibitors. Thus, it was encouraging to have a target that might lead to drugs with multiple potential applications. We also concluded that this effort was merited, since numerous past attempts to develop useful PNP inhibitors by standard trial and effort methods had not been successful. However, these past efforts had produced a number of inhibitors that, although not suitable for clinical use, would be available to us in our crystallographic work for characterizing the active site of the enzyme. With all of this in mind, John and I embarked on a path in the 1970's to undertake a project that would eventually cover many years of our future careers.
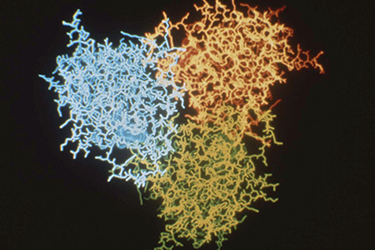
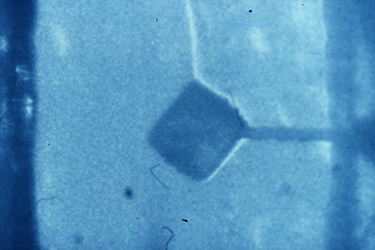
Much of the biochemistry of human PNP had been performed by Bob Parks and Johanna Stoeckler in the Department of Pharmacology at Brown University. They kindly agreed to collaborate with us on a crystallographic study of the enzyme structure, and they provided us with generous amounts of the purified enzyme isolated from human red blood cells. Bill Cook (Fig. 3) crystallized the enzyme at UAB in 1981 [91], and Steve Ealick then assumed the lead role in the crystallographic studies that eventually led to the structure of the enzyme [110, 139]. The crystallographic analysis was a fairly difficult undertaking at the time since the crystals had a very high 80% solvent content (Fig. 4), and thus diffracted relatively weakly. This very large solvent content later proved to be a blessing when preparing active site directed complexes of PNP for drug design studies, but the crystals were clearly good candidates for analysis using the newly available high intensity beam lines at synchrotron facilities. In 1981, I was eligible to take another sabbatical leave, and I returned to Oxford to assist Margaret Adams and her group complete the structural analysis of sheep liver 6-phosphogluconate dehydrogenase at 2.6A resolution [101]. At this stage, John Helliwell had completed his doctoral studies and moved to Daresbury in northern England where one of the newly constructed synchrotron facilities was available. John had developed a beam line for X-ray crystallography, and he was delighted to join us as a collaborator on the structural studies of PNP. John was joined in this effort by Trevor Greenhough, a bright and enthusiastic postdoctoral fellow in John's research group at the synchrotron facility. Steve Ealick came over to Oxford and then on to Daresbury to help collect the high-resolution diffraction data that led to the high-resolution structure of PNP. Trevor later moved to Birmingham to continue with this project, and John's collaboration continued for the years that it took to determine the structure and to characterize the enzyme substrate binding site by determining the structures of a number of complexes of PNP with substrate analogs and with inhibitors of the enzyme. While at Oxford during the 1981-1982 year, I was very fortunate to become close friends with Y.S. Babu (Fig. 5), who was a postdoctoral fellow working with Louise Johnson on the crystal structure of phosphorylase. Babu was generally regarded as one of the brightest crystallographers with the Oxford group, and I was immediately impressed by the long hours he spent in the crystallography lab. He was one of the few people working on weekends when I was able to get time on the new Evans and Sutherland computer graphics system which was used for interactively constructing protein tracings to fit electron density maps. Margaret and her students had succeeded at producing a high-resolution map of the enzyme, and I had taken on part of the responsibility of fitting the sequence to the electron density. Since we had obtained funds to set up a similar graphics facility in Birmingham, I was eager to learn as much as possible about its use, so I jumped at every chance to access the system when it was available for extended periods on weekends. Babu was often the only other person available at those times, and he was always generous in giving me help with my project. I ended up offering Babu a position with our group in Birmingham, and I was delighted when he agreed to join us and to help Steve Ealick and the others working on the crystallographic studies of PNP.
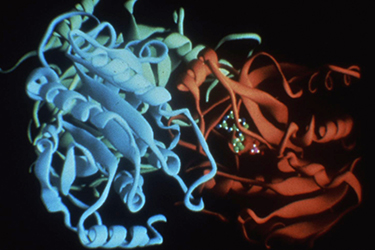
Steve Ealick led all of the crystallographic studies of PNP and of multiple complexes of the enzyme, work that encompassed much of the period between 1981 and 1985. He received tremendous help from other members of our crystallography group in Birmingham and from the Daresbury crystallography group, including Trevor Greenhough, Dan Carter, Steve Rule, J. Habash, and S.V.L. Narayana, along with continuous input from Babu, Bill Cook and John Helliwell. We continued to benefit greatly from continued collaboration with the biochemistry group at Brown University, including Bob Parks, Johanna Stoekler and S-F. Chen. By 1985, we felt that we had the structural data (Figs. 6 & 7) needed to begin a serious effort to design and develop useful inhibitors of PNP [112]. We initially applied to NIH for a program project grant to support the project which would require a fairly large effort involving crystallography, modeling, organic chemistry, biochemistry, and pharmacology just to get to a stage where we could adequately design, synthesize, and test inhibitors for preclinical development. Even with no funding requested for clinical development, the required budget for the initial project was huge. Our proposal received encouraging reviews, but the budget was judged to be beyond levels that NIH would consider for funding.
At this stage, we began to think seriously about seeking funding from private sources. Biotechnology was attracting considerable venture capital in the mid 1980's, and we were convinced that the PNP project had considerable long range commercial potential, especially considering the advanced stage of our crystallography, the extensive experience that John Montgomery had in medicinal chemistry of purine derivatives, and the multiple potential clinical uses for PNP inhibitors. We were fortunate to attract the interest of Bill Spencer, a local prominent business leader in Birmingham who was on the President's Council at UAB. Bill had started the first biotechnology company in Birmingham several years before, and he was enthusiastic about trying to help us raise venture capital to move forward with our PNP project. He had wonderful investor contacts in the southeast, and there were tax incentives in place at the time that made investments of this type especially favorable. In addition, UAB had very recently opened up The Center for the Advancement of Developing Industries, a startup company incubator on campus, and they were enthusiastic about having us as their first biotechnology occupant. After many presentations by me and John to qualified investors, primarily in the Birmingham area, Bill managed to raise $4.5 million dollars to start our venture. The prospectus that was made available to potential investors not only described the PNP project, but it also laid out our plans for future drug design projects directed at influenza neuraminidase, and at serine proteases, with an initial focus on complement enzymes. We came up with the corporate name of BioCryst Pharmaceuticals, Inc., after an evening of brainstorming over beer. Toward the end of our fundraising we were approached by Tom Glenn, who was director of research at Ciba-Geigy Pharmaceuticals (later acquired by Novartis) about a possible collaboration on the PNP project. Tom had been Chairman of Pharmacology at the University of South Alabama Medical School before moving to Ciba-Geigy, and he was familiar with our research activities in Birmingham. Tom was also interested in having his scientists learn more about structure-based drug design, and he appreciated the potential of PNP inhibitors as clinical candidates for treatment of autoimmune diseases. Ciba-Geigy agreed to make an investment in our new company and to dedicate two of their experienced scientists, Wayne Guida and Mark Erion, to work with us on the project. Wayne and Mark turned out to be really important additions to the PNP drug design team, since they brought pharmaceutical company experience, organization and discipline to our program. Babu became our first employee at BioCryst, which turned out to be one of the most productive recruitments I ever made in my career. Babu is a brilliant scientist with a tremendous background in protein crystallography, and he was totally familiar with the PNP crystallography and structure, having spent several years working in close collaboration on the project with Steve Ealick. Babu quickly became the heart and soul of BioCryst and turned out to be the future driving force in the design of multiple, exciting drug candidates at BioCryst over the years. Following guidance from our Ciba-Geigy colleagues, we established a drug design team consisting of Steve Ealick and Babu, working closely with Wayne Guida and Mark Erion from Ciba-Geigy, and John Montgomery and Jack Secrist who directed the organic chemistry efforts at Southern Research Institute (SRI). Crystallographic and modeling facilities were established at the UAB incubator, but the organic chemistry was subcontracted to SRI where they had extensive laboratory facilities that did not need to be duplicated immediately within the incubator. I Chaired a Scientific Advisory Board that was assembled for BioCryst, but I remained primarily committed to the various ongoing crystallography programs at UAB. I continued to follow the activities of the drug design team with great interest, but the crystallographic and design success of the PNP project over the following years were primarily due to the tremendous contributions from Steve and Babu, working with other members of the drug design team.
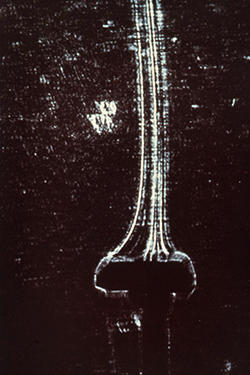
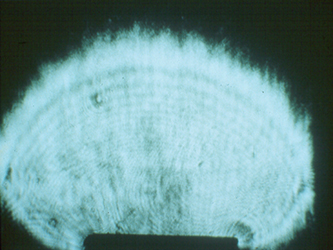
In 1985, our crystallography program at UAB took an unusual turn toward space. NASA was in the midst of designing the Space Station, and much of this work was being coordinated at the Marshall Space Flight Center in Huntsville, Alabama. The Space Station project was being driven by powerful political momentum in Congress. The contracts to construct the planned Station were to be divided among all fifty states. Consequently, the Space Station had broad support in Congress, who assumed that it would be put to good scientific use. NASA was actively in the process of trying to identify high priority scientific projects that could take proper advantage of this expensive facility. Our Alabama Senator Howell Heflin, who was from north Alabama and was a major supporter of the Space Station project, contacted the Director of the Marshall Space Flight Center and the President of UAB and urged them to get their two institutions together to identify projects in Alabama that might be competitive for funding from the Space Station science budget. I ended up on a committee from UAB to meet with the Huntsville scientists to see if we could identify common interests. I went reluctantly, since I had never thought about any possible ways our crystallography programs might relate to space. At that meeting, however, we were given a presentation about crystal growth experiments that had been performed years earlier in space, with very interesting results. These experiments involved optical measurements of disruptive convection caused by solution density changes during crystal growth, which were pronounced on Earth (Fig. 8) but were eliminated in microgravity (Fig. 9), resulting in enhanced quality of crystals grown in space. Crystal growth of electronic materials and metals had been identified as a priority area for microgravity research by the Marshall scientists. Mark Pusey, a member of the science team at the Marshall Spaceflight Center, later demonstrated that lysozyme crystals growing on Earth produced convective flow similar to that seen for other types of crystals (Fig. 10).
I presented some of our ongoing activities in protein crystallography at UAB and explained the challenges that everyone in our field was facing in the growth of high quality protein crystals for structural studies (Fig. 11). It quickly became obvious to the NASA scientists that protein crystal growth might be an incredibly important area for experiments in microgravity and for parallel NASA-sponsored research programs on Earth. It was clear that protein crystal growth had about every component they were looking for, including past evidence of microgravity effects on crystal growth, a key problem that plagued important crystallographic structural studies in biology, and potential commercial applications for structure-based drug design in the pharmaceutical industry. It was not clear how the effects observed in microgravity growth of inorganic materials might relate to growth of macromolecular crystals, but there was enthusiastic agreement that this was an area that should be investigated.
The Birmingham crystallography group immediately began working closely with scientists and engineers from the Marshall Space Flight Center to design suitable protein crystal growth experiments that might be performed on the Space Shuttle, which was flying on a fairly regular schedule at that time. We learned that an upcoming Space Shuttle mission was scheduled to include a biological experiment by McDonnell Douglas Aerospace Company using a large-scale electrophoresis system for purification of the protein erythropoietin, and one of the McDonnell Douglas engineers (Charlie Walker) would be going on the flight as a Payload Specialist to perform the experiment. After explaining our plans to the electrophoresis team, they agreed to take along our first protein crystal growth apparatus with them, and to have Charlie Walker perform the experiments. We only had a few months to design the experiments and the equipment that would allow us to take our first quick look at protein crystal growth in microgravity. Charlie Walker would have to store the crystal growth apparatus in limited space available among the electrophoresis equipment, and he would not have much time that he could allocate to our experiment, so this first experiment had to be fairly simple to perform. We quickly designed a simple vapor diffusion apparatus (Fig. 12) that could be activated in space with chambers to accommodate several different proteins. The apparatus was constructed in a local shop facility under the close supervision of NASA engineers, and our first microgravity protein crystal growth experiments were performed on Shuttle Flight STS-51D in April 1985. Vapor diffusion was selected as the first method to be explored since it was the technique most widely used for crystallization of proteins, and it allowed experiments to be performed using very small protein samples. Since it was the method of choice in protein crystallography, most of the data on quality of Earth grown crystals had been obtained using crystals grown by vapor diffusion, and crystallization conditions for many proteins were well developed using this method.
The vapor diffusion apparatus was formally designated by NASA as the Handheld VDA, and this designation was later extended to include an improved model developed after our first Space Shuttle flight. The initial Handheld VDA was composed of a series of cavities in an aluminum plate. A syringe containing the sample of protein solution was positioned at one end of the cavity, which contained an absorbent material soaked in the precipitant solution. At the other end of the cavity was a plunger that could be moved in and out to block or open the end of the syringe (Fig. 13). The cavities in the aluminum plate were covered by clear sheets of plastic on each side of the plate, so that the operations and crystal growth could be observed and photographed.
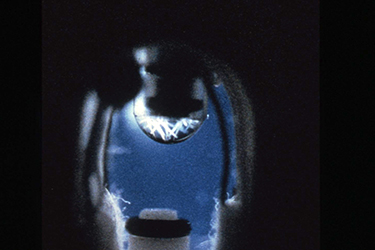
The protein solutions were prepared and loaded in the syringes at the launch site as close to the time of launch as possible, and the absorbent material was soaked with the solutions of precipitating agents prior to transferring the apparatus to the Shuttle. The system was then closed, and the plungers were positioned to seal the ends of the syringes. Both the syringes and the sealing plungers were operated by an Allen wrench. Bud Suddath, Larry DeLucas and I were responsible for loading proteins into the apparatus immediately before the Shuttle launch, using a laboratory that had been assigned to us at the Johnson Space Center (Figs. 14 & 15). The apparatus was then taken to the Shuttle and taped to the wall of a mid-deck locker assigned to Charlie Walker. Once in orbit (Fig. 16), Charlie removed the apparatus, and used an Allen wrench to retract the plungers from the syringes and then extrude the protein solutions onto the tips of the syringes. The crystallization process was then initiated by vapor equilibration between the precipitating agents and the droplets of protein solution. The droplets of protein solutions, along with microgravity grown crystals, would then be withdrawn back into the syringes, and the syringes would be capped by the plunger for return to Earth at the end of the Shuttle flight.
Depending on your point of view, this first experiment turned out to be either a valuable learning experiment or a gigantic failure. Out of the dozen or so samples, high quality crystals were obtained for only one protein, lysozyme (Fig. 17). There were major problems with stability of the droplets of protein solutions, and most of the droplets ended up splattered on the walls of the crystallization chambers. We concluded that unusual movement from firing of positioning rockets on that particular flight were probably a factor in the drop instability, and we decided that the syringe tips needed to be redesigned to add stability to the suspended droplets on future flights. In fact, the one successful experiment on STS-51D, which produced the large lysozyme crystal, used a different type of flared syringe tip that Bud Suddath added at the final stages in the event that we did experience unexpected movements. At any rate, we felt that we learned what we needed to know for designing an improved apparatus for future microgravity experiments by vapor diffusion. We also learned a tremendous amount about dealing with NASA, the facilities available for sample preparation at the launch site, and the procedures that had to be followed for developing equipment that would be acceptable for use on the Shuttle. We immediately began working on modifications to the Handheld VDA for future Space Shuttle flights. Meanwhile, there was much debate in the crystallographic community about whether or not any of this was really worth pursuing. Following our experiments on STS-51D, Gina Kolata published an article in Science titled, "The Great Crystal Caper" describing our first microgravity experiment in unflattering terms, along with a discussion of the ongoing debate among crystallographers about the merits of trying to grow protein crystals in space [21]. Our colleagues inside NASA, however, remained highly committed to moving this program forward. Protein crystal growth had everything they were looking for in a worthwhile experimental program that could be pursued initially on the Space Shuttle and eventually on the Space Station. Protein crystallography had huge science appeal and was rapidly being implemented in drug design programs within pharmaceutical companies around the world. Crystal growth was a major problem in protein crystallography, and anything that potentially might improve the process would be important. Finally, there were reasonable theories about why microgravity should affect crystal growth processes, and previous microgravity experiments with growth of inorganic crystals had yielded data supporting these theories. All of this was easy for top administrators within NASA to appreciate and for Congressional funding committees to understand. Microgravity protein crystal growth was here to stay, at least until enough experimental results had been accumulated to conclude it was a complete waste of further effort. Working closely with NASA engineers, we modified the Handheld VDA trying to incorporate changes that would maximize solution stability and better control the vapor diffusion process. This improved Handheld VDA was flown on two additional Shuttle flights in 1985, each time showing us additional improvements that could be made to the apparatus and the processes involved. On these other 1985 Shuttle flights, we never really had much time that the crew could give to our experiments, since their schedules were tightly controlled by activities that had been planned years in advance.
Our first real opportunity to have a dedicated person on board for our experiments came with STS-61C, in January 1986. For this set of experiments, we had a fully devoted Payload Specialist, Congressman Bill Nelson from Florida, to perform the flight experiments. Congressman Nelson (now Senator Nelson) was on one of the House committees that oversees NASA, and he arranged to go on the Shuttle flight as part of his oversight responsibilities. Congressman Nelson was able to select an experiment that he could help perform on the mission. He enthusiastically chose our protein crystal growth experiments, largely because of the linkage of protein crystallography to important biomedical research. He was a wonderful person to work with. He trained with Larry DeLucas with our crystal growth apparatus on the KC-135 airplane (better known as the "Vomit Comet") that NASA used to generate brief periods of low gravity by flying parabolic patterns (Fig. 18). He also spent several extended periods in Birmingham training on prototypes of our crystal growth apparatus, asking lots of detailed questions and getting totally familiar with the procedures that would be involved. He successfully completed our experiments on the Shuttle flight, which launched on January 12, 1986 (Fig. 19). The results were again somewhat mixed but considerably improved over those from that first experiment. Congressman Nelson became a major advocate in congress for protein crystallography, and he was awarded the Public Service Award at the Philadelphia ACA meeting in 1987, at which I retired as President of the organization. He also wrote a book titled, "Mission" that described his brief career in protein crystallography and the experiments he performed for us in space. Our experiments with Congressman Nelson were on the last Shuttle flight prior to the Challenger disaster on January 28, 1986. We did not have experiments on Challenger. Following the Challenger accident there was a long extended period when the Space Shuttle program was grounded, while NASA implemented changes to enhance the safety of the program. During this period we worked closely with the NASA engineers to develop an Advanced Vapor Diffusion Apparatus (known in NASA as the Advanced VDA) that incorporated changes to take advantage of all that we had learned from the four flights with the Handheld VDA. This allowed many more experiments to be performed simultaneously. This hardware was automated, so it could be operated with minimal crew time and training. The Advanced VDA was enclosed in a temperature-controlled unit designed to replace one of the mid-deck lockers on the Space Shuttle. The Advanced VDA (Fig. 20) included sixty crystal growth chambers and greatly improved the control of crystal growth conditions on future flights. Using the Advanced VDA, we performed experiments on eight more Shuttle flights between the Challenger accident and the time when I retired from UAB.
Figure 20: Payload specialist Pinky Nelson (no kin to Congressman Nelson) shown training with the Advanced VDA, which was constructed following the Challenger accident. This system was more automated, and the vapor diffusion apparatus was housed in a special temperature-controlled unit that was designed to replace one of the mid-deck lockers on the Space Shuttle. Pinky performed our first experiments with the Advanced VDA on Space Shuttle Flight STS-26, which launched on September 29, 1988. Shortly after our first microgravity experiments, NASA initiated a program to establish a series of university based centers to pursue technologies that could benefit from space access, with particular emphasis on technologies that had commercial potential. These Centers for Commercial Development of Space (CCDS) were to be spread across several technologies and areas of science that might eventually enhance the commercial potential of space. We felt that protein crystal growth and various applications of protein crystallography came as close to meeting that goal as any other discipline. We were heavily involved at that stage with structure-based drug design program in BioCryst, and we had several outstanding protein crystallographers from pharmaceutical companies collaborating with us on microgravity protein growth experiments. We applied for a Center grant to provide broad support for our crystallography programs in Birmingham and for collaborative studies with commercial partners. We were funded generously by NASA in the first round of applicants, and the Alabama Board of Trustees approved formation of our new Center for Macromolecular Crystallography (CMC). I was the initial Director, Bud Suddath was Associate Director for X-ray Crystallography, and Larry Delucas was Associate Director for Protein Crystal Growth. Several pharmaceutical companies who had been collaborating with us became commercial partners in the Center. The Center designation gave us considerable independence within the university, which facilitated future expansion of our crystallography program in Birmingham. The CMC also served NASA as the conduit through which our many collaborators participated in microgravity protein crystal growth experiments, as Members of the CMC. The microgravity program included an outstanding group of collaborators from academia, government labs and industry. At the time that I left UAB in 1994, we had an international group of collaborators from 23 universities, 12 pharmaceutical companies, and 4 government laboratories working with us on microgravity protein crystal growth experiments. One particularly valuable collaboration that grew from our early space experiments was with Schering Plough Pharmaceutical Company (later acquired by Merck). At that time, Dr. T. L. (Nag) Nagabhushan, who was Senior Vice President of Research at Schering Plough, was heavily involved in productive biochemical and clinical studies of interferons, protein growth factors, cytokines and lymphokines, and he was eager to pursue structural studies of these interesting proteins. Alpha interferon was a major pharmaceutical product for Schering Plough, and their manufacturing process was producing large quantities of highly purified protein for marketing. Therefore, this well characterized protein was available in the large quantities that might allow us to expand the microgravity experiments beyond the vapor diffusion studies designed to accommodate milligram samples of protein. Considering the evidence that microgravity may affect crystal order, it seemed possible that microgravity crystallization might provide a route for enhanced large-scale purification of biologicals, as a step in the final manufacturing of commercial protein products. This concept was very appealing to the commercialization division of NASA, since there were very few products that could possibly justify the high expected costs of manufacturing in space. If large-scale microgravity crystallization of biologicals did enhance the quality of the final product, then the high price that biologicals generate per gram of weight might eventually justify the high cost of manufacturing protein products in space. This same rationale was the basis of the electrophoresis experiments that Charlie Walker had performed for McDonnell Douglas with erythropoietin on STS-51D, when we piggybacked our first microgravity protein crystal growth experiments on his protein purification apparatus. NASA understood this rationale well, and our microgravity protein crystal growth program picked up a new burst of momentum as we expanded into large-scale protein crystallization studies. In collaboration with Nag's group at Schering Plough, we proceeded to design equipment for conducting microgravity bulk crystallization experiments. The system we ended up designing used temperature change to activate crystallization, which is one of the most common techniques used in manufacturing of pharmaceuticals. This also was a fairly straightforward method for scaling up our experiments to accommodate large samples. Within the CMC, these experiments were coordinated by Marianna Long, who worked closely with various members of Nag's group at Schering Plough. Temperature change is potentially ideal for optimizing microgravity effects, since temperature driven convection is essentially eliminated in space along with normal crystal growth convection and crystal sedimentation. The system designed for these bulk crystallization experiments was fairly simple, consisting of clear plastic cylinders of various volumes with metal caps that abutted a metal plate that had precise temperature control. The cylinders were loaded with protein solutions prior to launch and were maintained at temperatures where the proteins were soluble. Once in orbit, the temperature of the solution was gradually adjusted by changing the temperature of the metal plate that was in contact with the metal cap of the cylinder. This resulted in a temperature gradient across the cylinder, which would have been unstable in the convection driven flows on Earth, but was quite stable in microgravity. The temperature across the solution then gradually equilibrated during the flight, producing an unusually stable environment for gradual nucleation and growth of large quantities of crystalline material. The rate of equilibration could be controlled by varying the rate of temperature change on the temperature control plate, depending upon the length of time that the Shuttle was expected to remain in orbit. This bulk crystallization system (known within NASA as the Protein Crystallization Facility, or PCF) was tested on four Shuttle flights between 1991 and 1994, using samples of bovine insulin, alpha interferon and human insulin. The results were encouraging, producing crystals in microgravity that were generally considerably larger and better formed that those grown in this system on Earth. In addition, crystals of human insulin appeared to diffract to significantly higher resolutions, suggesting enhanced internal order. Probably the best-characterized crystals obtained in these initial bulk crystallization experiments were those obtained for a human insulin complex by Dave Smith and his colleagues at the Hauptman Woodward Medical Institute. They reported obtaining crystals from their microgravity experiment that were larger and of significantly higher quality than crystals grown in normal gravity. Using six of these microgravity grown crystals for data collection, the Buffalo group successfully refined the insulin complex structure to 1.4 Å resolution, including hydrogen atoms in the final refined model. Protein crystal growth soon became a major program within NASA, and they provided grants to several academic groups to pursue a variety of projects to better understand this important field on Earth. A protein crystal growth research program was established at the Marshall Space Flight Center, and we expanded our program in Birmingham to help coordinate microgravity protein crystal growth experiments with numerous collaborators from other universities and from the pharmaceutical industry. Larry Delucas became our key leader in Birmingham for coordinating our space activities, and we were soon performing experiments on most Shuttle flights. Meanwhile, the demands on my time from NASA became more and more overwhelming, especially as planning for the construction of the Space Station picked up momentum. It was assumed within NASA that protein crystal growth would be a major program on the Space Station, and facilities to accommodate that program were being included in all of the early stage planning for the Station. The tentative plans even included serious discussion of including an X-Ray diffraction facility onboard the Space Station so that crystals could be characterized quickly, and complete data sets might be collected without waiting for return to Earth. I ended up as Chairman of the Biotechnology Discipline Working Group of the Universities Space Research Association; a Member of the Committee on Industrial Applications of the Microgravity Environment, Space Applications Board, National Research Council; and a Member of the NASA Space Station Science and Applications Advisory Subcommittee. Since the Space Station was an international effort, I was called on often to meet with science groups from Europe and Japan. Beginning in 1985, I was Principal Investigator of the large, and rapidly expanding NASA contract to support the microgravity program in protein crystal growth, including funds for all of the space activities; for construction of special equipment for microgravity experiments; for research in our Center to better understand factors that affect crystal quality; for coordination of the rapidly expanding international group of collaborators who were steadily joining the program; and for the hiring and management of engineers and other personnel required to meet the NASA demands for huge quantities of paperwork. Our key personnel working on the program were screened and given high level government clearance that would allow them to have ready access to NASA facilities, and we established laboratory facilities at the Kennedy Space Center for preparing experiments close to the launch site. The research group at the Marshall Space Flight Center also grew to include protein crystallography and basic programs in protein crystal growth. I was spending a huge part of my time flying from one NASA meeting to another, including many in Europe and Japan, while I really needed and wanted to concentrate on our other protein crystallography programs, the exciting protein structural studies we had underway, and our fruitful collaboration with BioCryst in structure-based drug design. Fortunately, Larry Delucas was completely dedicated to our NASA program, and he was soon heavily involved in coordinating all aspects of the program. He had the scientific background and the personality to smooth over the multiple bumps that confronted us daily and to interface effectively with our many colleagues in NASA and did a wonderful job overseeing all of our space experiments, supervising design of our evolving protein crystal growth equipment, training Payload Specialists to perform our Shuttle experiments, and interacting with the program managers within NASA.
As the microgravity Space Shuttle experiments advanced through a series of Shuttle flights, data accumulated in support of the theory that more highly ordered protein crystals could be grown under microgravity conditions. The most convincing data came from careful analysis of diffraction intensities as a function of scattering angles when comparing Shuttle grown crystals with control samples. The data that I found most helpful in evaluating crystal order was based on the types of relative Wilson plots (Fig. 21) that had been used historically to scale intensity data from heavy atom derivatives of protein crystals to data from the native protein crystals, to compensate for the disordering effects that the derivatives often induced in the native crystals. Similar disordering effects were apparent in relative Wilson plots comparing Earth grown and microgravity grown crystals (Fig. 22), indicating that crystals grown in microgravity tended to be more highly ordered, with measurable diffraction data that extended to higher resolution [132]. However, debates continued in the crystallographic community about how worthwhile this effort really was and if the funding required might be better used for other purposes. I was sympathetic to the funding question, since we had other projects supported by NIH, but the reality was that the funding that Congress awarded to NASA was somewhat independent from that allocated to NIH. Microgravity protein crystal growth picked up great momentum in the late 1980's, and there was building interest in upcoming opportunities to perform experiments over longer periods of time, first on an upcoming Spacelab mission that was planned for 1992, and later on the Space Station. The Spacelab mission might also be the first good opportunity to get a qualified crystallographer on board the Space Shuttle, since Spacelab flights are fully dedicated to science. One advantage of all the NASA committees that were soaking up my time was that I was in a strong position to effectively argue for a science slot for a crystallographer on the planned Spacelab mission. If NASA agreed that a protein crystallographer should be included among the scientists on the Spacelab mission, the most logical person to fill that role was Larry DeLucas, since he had the experience and credentials to be a perfect choice. In 1991, Larry was selected to fly as a Payload Specialist on STS-50, and he began a year of training for the job. His mission was launched on June 25, 1992 (Fig. 23), and it gave us the longest uninterrupted period we had ever had to do experiments before the Shuttle returned on July 9 with Larry and the crystals in great shape.
Figure 23: Larry DeLucas performing protein crystal growth experiments on Spacelab STS-50. This was a fifteen-day mission, which gave us our longest period of uninterrupted protein crystal growth in microgravity. Before I left the microgravity protein crystal growth program to join BioCryst in 1994, we had performed experiments on sixteen Shuttle flights [114, 116-118, 122, 128, 130, 132, 133, 140, 142-147, 154, 159, 165, 169]. Four of these were performed with early stage versions of equipment for vapor diffusion experiments (the Handheld VDA). The others were performed using the Advanced Vapor Diffusion Apparatus that was developed post Challenger (the Advanced VDA), and the batch crystallization facility (the PCF). Eight of the post Challenger flights included vapor diffusion experiments, and four included large-scale crystallization experiments using the thermal gradient crystal growth equipment. A total of 81 different proteins, provided by some forty collaborators from protein crystallography groups around the world, were included in crystal growth experiments on these sixteen Shuttle flights. Of these, fewer than twenty percent produced microgravity grown crystals that were noticeably improved over those obtained on Earth. Many of these vapor diffusion experiments included proteins that were very poorly crystallized on Earth in hopes that space might provide a chance for obtaining useful crystals from proteins that had proved intractable in crystallization experiments on Earth. These "Hail Mary" protein crystal growth experiments almost invariably failed to yield positive results in space. The most encouraging results were obtained in the space experiments with proteins that had been studied extensively, with successful crystallization results already obtained on Earth. Among this subset of proteins, there were several striking examples of improved crystal order as evidenced by enhanced diffraction resolutions and reproducible data from relative Wilson plots [132]. I have not closely followed the many microgravity experiments since 1993 that the Birmingham group has coordinated on Shuttle flights, and more recently on the Space Station, but the results that I have seen from recent reviews support our earlier conclusions that significant improvement in crystal quality may be obtained in space if the samples included in the experiments are carefully selected from extensive Earth experiments prior to space flight. At the time of this writing (January, 2015) a huge set of protein crystal growth experiments has just recently been returned from the Space Station for analysis by Larry and his collaborators, to evaluate the long-range potential of microgravity protein crystal growth. The proteins included have all been crystallized previously on Earth. The experiments are double blinded, with Earth experiments being performed in the same type of equipment, in parallel with the Space Station experiments. The crystals will all be analyzed by various standardized diffraction techniques in Birmingham. The diffraction analyses will be blinded experiments that will be used to compare order of the space grown crystals with their earth grown counterparts. These analyses are expected to take the better part of the next year, and the results are expected to be important in helping to determine the future direction for microgravity protein crystal growth. Our collaboration with Nag's group at Schering Plough evolved into a broad and expanding program in crystal structure studies of lymphokines and cytokines. Schering Plough did not have a crystallography program at the time, and our initial crystal growth collaboration with Nag and his group soon expanded to include multiple structural studies of the exciting proteins that they were developing for potential commercial applications. Bill Cook worked closely with Nag's group on crystallization of these proteins, and Steve Ealick led the crystallographic work that ended up determining the structures of human gamma interferon [120, 153], human granulocyte-macrophage colony stimulating factor (GMCSF) [137, 155], and human interleukin-4 [149, 156]. Mark Walter worked closely with Steve Ealick and Bill Cook on these crystallographic studies, and he carried on the collaborative program with Nag and his group following my departure from UAB. This work led to the 1994 Milstein Award for Interferon and Cytokine Research given to me and Nag as representatives of the UAB and Schering Plough groups. These structural studies had only been possible because of the major contributions from Steve Ealick, Bill Cook, Mark Walter, and Vijay Senadhi at UAB; and Paul Trotta, Paul Reichert, G.S. Hammond, and H.V. Le at Schering Plough. Nag and I considered ourselves to simply be representatives of this superb group of crystallographers and biochemists in accepting the award. The collaboration with Schering Plough also produced a beautiful new artistic representation of protein structure carved in wood. Academic groups could never have undertaken such a project, but the interferons and lymphokines were major commercial products for Schering Plough, and they were eager to display our crystallographic results in the entrance to their new research facility, which was scheduled to open in New Jersey soon after we had completed the structure of gamma interferon. Bebe worked for a wonderful art gallery in Birmingham at the time, which specialized in sculptures done in various materials, and she was familiar with the spectacular work that a local artist, Craig Nutt, was doing with wood carvings. Bebe and I met with Craig, who knew nothing about molecular structures, and we showed him some of Mike Carson's computer graphics representations of the gamma interferon structure. He was blown away by the complexity but was awed by the beauty of the interlacing helices, which are abundant in the interferon structure. He gave us a rough estimate of the time and dollars that would be required to fabricate an accurate wooden carving of the structure. I thought there was no way Schering Plough would be willing to fund the venture, considering the price, but they approved it without hesitation. Craig and Mike Carson then spent many hours together with our graphics system before Craig was comfortable beginning his carving. The results are spectacular (Fig. 24). The final model depicts the complete backbone tracing, carved in cherry wood, with dimensions that scale almost perfectly with the actual structure. The final size is about 10ft x 7ft x 5ft, and the carving was available to adorn the new Schering Plough facility shortly after it opened. Craig named this wonderful artistic representation "Helical Dance." I have not seen anything like it since, and I doubt if the intricate style will be duplicated for many other protein projects.
Figure 24: The beautiful model of gamma interferon, carved in cherry wood, which was commissioned by Schering Plough Pharmaceutical Company to adorn the lobby of their new research center in New Jersey. T.L. (Nag) Nagabhushan is pictured on the left, and the artist, Craig Nutt, is shown on the right. Craig is holding a small scale carving of the structure, which he completed before embarking on the larger carving. The molecule is carved nearly perfectly to scale, thanks to the collaboration between Craig and Mike Carson. Protein crystallography made major progress during the 1980's, and the rate at which new protein structures were being determined seemed increasingly reminiscent of the growth that I experienced in small-molecule crystallography during the 1960's and 1970's. When I did my PhD thesis work in the early 1960's, determining the structure of a small organic molecule was a major challenge, but all new structures seemed to reveal something of fundamental importance for understanding chemical bonding and molecular interactions. By the late 1980's, many thousands of small-molecule structures had been determined and were deposited in the Cambridge Structural Database. Thanks to advances in direct methods of phasing, and automated data collection methods, determining the crystal structures of most small molecules had become somewhat routine, and most chemistry departments had established crystallography facilities for structure analysis by non-specialists. By the 1980's, I felt I was in the midst of a similar revolution in protein crystallography even though the Brookhaven Protein Data Bank only contained a few hundred structures at the time. Each new protein structural study still was a major challenge, but synchrotron facilities and newer phasing techniques held the promise of rapidly increasing the rate at which new protein crystal structures would be determined in the near future. The 1980's were an incredible period to be in protein crystallography since almost any new protein crystal structure seemed to reveal important new information in structural biology. However, I had an increasingly uneasy feeling that our unique talents as protein crystallographers were rapidly transitioning from unique to more routine. In 1987, I had the pleasure of serving as the President of the American Crystallographic Association, and I decided to focus on the future of protein crystallography for my after-dinner talk at the Philadelphia ACA meeting. I showed plots of the past growth of the Cambridge Structural Database and of the current growth rate of the Brookhaven Protein Data Bank, and I suggested that the plots overlaid pretty nicely when comparing the early stages of small-molecule crystallography with the then current growth rate for new protein crystal structures. If we assumed that the two growth functions were going to be approximately the same, I suggested that we could reasonably expect thousands of new protein crystal structures to be forthcoming during the next few years. This suggestion was met with considerable skepticism from my colleagues, but it really started me thinking about how I wanted to spend the rest of my research career. I had lived through the upheaval in small-molecule crystallography when my special crystallographic expertise transitioned from special to routine, and I had the eerie feeling that we were not far from the time when protein crystallography would also become a tool widely adopted by non-specialists. Obviously, this would be great for science, but it could potentially affect the role of crystallographic specialists in the future. My thoughts about the future of protein crystallography were further enlightened by an unusual event that occurred while I was serving as a protein crystallography reviewer on the NIH Molecular and Cellular Biophysics Study Committee in the late 1980's. We had received an interesting grant proposal from a molecular biologist who had no experience in crystallography, but who was proposing to determine the structures of proteins that were of central importance in his research program. He recognized that structure was of central importance in understanding the biology of his systems, and he did not want to depend on someone else for this experimental procedure. He had arranged with his university to take a sabbatical and to work closely with the protein crystallographer at his medical school, and he was determined to learn what he needed to know so that he could use protein crystallography in his molecular biology research program. At the time, it was heresy to think about funding a protein crystallography project of a non-crystallographer, but this scientist was Hamilton Smith who had previously received a Nobel Prize for his research on restriction enzymes. Dr. Smith was at Johns Hopkins Medical School, and Ed Lattman, a superb protein crystallographer there, had agreed to serve as his tutor. We felt it would be a mistake for our Study Section to reject the proposal without further analysis, and I was appointed Chairman of a site visit committee from our Study Section to go to Hopkins and evaluate the research proposal. That site visit was eye opening for me. After spending the day with Dr. Smith, I was personally convinced that protein crystallography was on the path of becoming a major tool that would be incorporated in molecular biology programs, much like small-molecule crystallography had become a major tool in chemistry departments. Protein crystallography would likely remain a major area for theoretical and experimental research by card carrying crystallographers, especially when really challenging structural studies were involved, but I could see the day was rapidly approaching when our discipline would also transition to the hands of other scientists who used it as a major tool in advancing their research in structural biology. Experienced crystallographers were likely to be in increasing demand as the field of structural biology grew, but it seemed likely that more and more non-crystallographers would be adopting crystallography as an essential tool in all areas of molecular biology, biotechnology and pharmaceutical research. I began to question where I wanted to fit into this scenario for my future career, and I became increasingly convinced that structure-based drug design was an attractive option. If protein crystallography were going to advance as rapidly as I expected, it was clear that the crystallographic community needed to prepare for how we were going to handle a surge of new structural data, which would likely challenge our databases and journals. I had seen firsthand how powerful and useful the Cambridge Structural Database had been for making small-molecule structural data widely available, and I knew a little about the tremendous infrastructure and staffing that had been expertly assembled in Cambridge to handle the huge flow of small-molecule crystallographic data. It seemed likely that the Brookhaven Protein Data Bank, which was the protein structure depository at the time, would soon face a similar rapid increase in demands from the crystallographic and biological communities to handle a dramatic increase in protein structural data. During the 1988-1994 period, I was a Member of the Visiting Committee in Biology at the Brookhaven National Laboratory, and I was also appointed Chairman of the Brookhaven Protein Data Bank Advisory Board in 1992. Our Advisory Board was in a position to evaluate how ready Brookhaven was to handle the growth that we expected, and we felt obligated to do all we could to help Brookhaven adequately prepare for the anticipated influx of new data. Tom Koetzle, who was Director of the Data Bank at the time, was heavily committed to the program and was doing a yeoman's job operating this important national asset with a very small staff. Our Advisory Board strongly felt that he was greatly underfunded and understaffed to cope with the future, and we made our opinion clearly known to the upper management at Brookhaven. I felt we were somewhat successful at helping Brookhaven management understand the importance of the facility and the need to increase the funding and staffing that would be required to meet future demands. We also helped raise the issue to the national level of attention, and eventually NIH and NSF stepped in to add their influence and funding to help establish first class facilities for assuring that protein structural data would be widely available to the international community. Unfortunately, Brookhaven did not end up as one of the final Protein Data Bank facilities. The last time I looked, the Protein Data Bank, which is now managed at Rutgers University and The University of San Diego, has data for well over 100,000 protein structures and is still growing rapidly. I also had the pleasure of serving as Editor-in-Chief of Acta Crystallographica and the IUCr Commission on Journals during the 1987-1996 period (Fig. 25). By the latter part of my term as Editor ofActa Cryst., protein crystallography was expanding rapidly, especially as biochemistry and biology departments established crystallography groups to participate in the growing field of structural biology. Our understanding of protein structure was still in the infant stage, and almost every new protein or nucleic acid crystal structure was of great interest to the structural biology and molecular biology communities. Consequently, most of the macromolecular crystallographic results were published in biochemical and biological journals. These journals were understandably primarily interested in the biological implications of the resulting structures, rather than the crystallographic details that made the structural results possible. The IUCr Executive Committee and the Commission on Journals became increasingly concerned that we were not adequately serving the biological crystallography community, which was growing rapidly at that time. There was clearly a need for a journal that could cover the many aspects of protein crystallography that were essential for development of the field, including key theoretical studies in protein crystallography; new methods and facilities for data collection and processing; advances in protein crystal growth theory and procedures; methods for evaluating the accuracy of structural results; modeling methods for fitting proteins to electron density maps and refining the structures; new approaches to phasing; and details of biological structures that might be of little immediate interest to biologists. Many details that were likely to be of importance for future advances in the nuts and bolts of macromolecular crystallography needed to be published even though these details might be of little interest to readers of biological journals, and we felt thatActa Crystallographicashould address this opportunity. After much discussion with the protein crystallography community, and with the enthusiastic support of André Authier,President of the IUCr at the time, we initiatedActa Crystallographica, Section D, titled "Biological Crystallography," which is now one of the most popular journals in theActafamily.
Figure 25: My final meeting with the Commission on Journals and the Executive Committee of the International Union of Crystallography (IUCr) at the IUCr meeting in Seattle in 1996. Shown from left to right, are Syd Hall, Mike Glazer, Samar Hasnain, me, John Helliwell, André Authier and Philip Coppens. John Helliwell and I spent many years collaborating on various crystallographic projects, and he succeeded me as Editor-in-Chief of Acta Crystallographica.Samar Hasnain followed John in that position. André retired as President of the IUCr at the Seattle meeting, and was succeeded by Philip Coppens. André worked very closely with me in initiating Acta Crystallographica, Section D, Biological Crystallography. During the late 1980's, our crystallography group at UAB became increasingly focused on structure-based drug design [135, 141], and we initiated crystallographic studies of several additional enzymes that we felt would be especially suitable drug design targets, including influenza neuraminidase, which was being pursued by Ming Luo and his students, and complement proteins [150, 163, 167], which were being pursued by Larry DeLucas and N.V.L. Narayana in collaboration with John Volanakis from the UAB Department of Medicine. Both of these programs were later licensed from UAB to BioCryst. UAB was also focused on new approaches to molecular modeling that might be of broad use in structure-based drug design. Mike Carson led a creative modeling program focused on novel approaches for displaying protein sites by computer graphics in ways that would allow non-crystallographers to see features that would be helpful in drug design [134,136]. Mike's early work produced the now popular algorithm for ribbon representation of polypeptide chains [113, 168], and he designed new ways of displaying and interacting with protein sites [152]. Scott Rowland pioneered other creative approaches for predicting interaction patterns that might be applied to drug design through extensive analysis of intermolecular contacts found in small-molecule crystal structures from the Cambridge Structural Database. Scott later extended these studies as a member of the Cambridge Structural Data Center staff, followed by several years at BioCryst as a member of our drug design group. By 1993, our BioCryst/Ciba Geigy/UAB/SRI collaboration had produced a series of potent inhibitors of human PNP [151, 158, 160-162, 166, 170], and a lead candidate, BCX-34 (later assigned the generic name peldesine) had been selected for clinical development by BioCryst (Figs. 7 & 26). A second PNP inhibitor, BCX-5 (Fig. 26), was partnered with Warner Lambert Pharmaceutical Company for clinical development. When John Montgomery and I originally selected the PNP target for drug design back in the late 1970's, the objective was to end up with drugs for treating patients, so we were finally at an important milestone. The challenge we faced at that stage was to come up with the funds necessary to move BCX-34 forward into clinical development. I ended up grossly underestimating how much it would eventually cost to develop a PNP inhibitor, but it was clear that we would need to raise a lot of money to even initiate clinical development properly. Between 1986, when we first incorporated BioCryst, and 1993, we had repeatedly gone back to our original investors to raise additional funds. We had also brought in funding from a couple of prominent venture capitalists from national investment firms. However, these investors were not willing to undertake the complete costs that would be required for clinical development of BCX-34, along with our planned expanded program for attacking additional targets. Our investors were painfully aware that drug development is incredibly expensive, very risky with high failure rates, and takes a long time to complete the necessary clinical trials for drug approval by the FDA. It was going to take a lot of capital, available continuously over a number of years, if we were to realize the goal of making our PNP inhibitors and other compounds available for treating patients. The ideal strategy for us was to take BioCryst public through an initial public offering (IPO) of stock in the company. A publicly traded company has much better access to investors than we would ever have as a private company, since stock in the company would then be a liquid asset that could be bought and sold on public markets. Also, the business aspects of the company would then be transparent and continuously monitored by the Securities Exchange Commission, thus allowing large investors and pharmaceutical partners to be confident that they could adequately evaluate and monitor their investment. I knew almost nothing about the details of how we would go about an IPO process, but the primary venture capitalists who had invested in BioCryst at the time knew what needed to be done, and they had the Wall Street contacts who might be helpful in moving the process forward. With their help and guidance, I began participating in a series of frequent trips to New York with the CEO of BioCryst and meeting with investment bankers and analysts to tell the BioCryst story. By late 1993, we had two top-tier investment banking firms that wanted to represent us in an IPO, and both firms had superb technical analysts who were convinced that PNP was an exciting target and that structure-based drug design was a promising technology. Our case was especially reinforced by an invitedScientific Americanarticle on structure-based drug design by John Montgomery, Mike Carson and me [164] which was published at the same time we were in the process of discussing an IPO. The New York bankers and analysts don't usually readActa Crystallographica, but they do followScientific American, and the issue with our article was on the newsstands in New York in late 1993 while we were actively trying to put together the IPO. Even though the stock market was beginning to soften at that time, and biotechnology IPOs were rapidly falling out of favor, it appeared that BioCryst could successfully complete an IPO, but with one big condition. The bankers, analysts and the major investors involved felt that it would be critical for me to leave UAB and go fulltime with BioCryst. John Montgomery had already left his long time position at SRI and taken over the medicinal chemistry program within BioCryst, but our Wall Street colleagues argued that it would be essential for me to also join the company, if they were to be successful at completing the IPO. I had never even considered leaving UAB and joining BioCryst. Even after John Montgomery made his move to BioCryst, I assumed that we could continue the collaborative program that allowed me to remain at UAB. I had been at UAB since 1968. We had a robust crystallography program at UAB where I had a superb group of graduate students, postdoctoral fellows and other colleagues. We also had been very successful at obtaining grants from NIH, NASA, non- profit agencies, and pharmaceutical companies; and our crystallography programs were going great. I was very happy at UAB and planned to stay until I either died or retired at an old age. I was a tenured Professor in the Department of Biochemistry and Molecular Genetics and was presumably set for life. On the other hand, it was important to me that BioCryst succeed. I had convinced friends, family and important people around Birmingham to invest in the company over those years between 1986 and 1993. I believed strongly in what BioCryst was doing, in the potential of PNP inhibitors, and in the future of structure-based design. However, I realized that the future of the whole BioCryst program was in jeopardy if we did not take this opportunity to complete an IPO and raise the necessary funds to move forward. Bebe was supportive of whatever decision I came to, even though leaving the security of my position at UAB was a risky move and would likely turn out to be a life altering event for our family. Bebe probably would have vetoed the move if Penny Mann, my wonderfully proficient administrative assistant at UAB, had not agreed to leave the university and come along to keep me organized, but fortunately Penny did. In November 1993, I formally notified the UAB administration that I planned to retire at the end of the year and take over as the Chairman and CEO of BioCryst. At that time, the Center For Macromolecular Crystallography had major funding from NASA, and Larry DeLucas was highly qualified to succeed me as Director of the CMC. The Department of Biochemistry and Molecular Genetics generously offered me an ongoing position of Professor Emeritus following my retirement. My contract with BioCryst was written to allow me to continue spending up to 10% of my time with UAB, which allowed me to keep my grants active until they could eventually be transitioned over to other principal investigators. I was honored to be selected the 1993 Distinguished Faculty Lecturer for UAB (an election that was made before I announced my impending retirement) which gave me a wonderful opportunity at a formal dinner in late November to say farewell to my UAB friends and colleagues and to describe what we had planned for the research programs at BioCryst. So on January 1, 1994, I jumped from my secure academic nest into the corporate world of biotechnology. It was immediately clear that I had a lot to learn, and I needed to learn it quickly. I had actually been fairly insulated from the internal operations of BioCryst while I was at UAB, and I knew very little about the company beyond the drug design programs. Since I had been Principal Investigator on several grants at UAB that overlapped BioCryst activities, and UAB had license agreements with BioCryst and equity in the company, the University had been careful to minimize any potential conflicts of interest between me and BioCryst. Consequently, we had set a fairly clear boundary between me and the company while I was at UAB. I chaired the BioCryst Scientific Advisory Board, which was primarily focused on structure-based drug design and selection of promising new potential targets for BioCryst to pursue, but I had minimal involvement in other BioCryst activities. I had never even met the Chief Financial Officer, the Vice President of Clinical Development or the Vice President of Regulatory Affairs, key people who would now be responsible for financial management and for initiating and overseeing the BCX-34 clinical development program. I also had limited knowledge about the business activities that BioCryst had been pursuing over the years since we raised our original funds from investors. During the first three months of 1994, I was focused heavily on the IPO, which was the major carrot that had lured me from UAB. During this period, I spent more time in New York than in Birmingham dealing with investment bankers, analysts, lawyers, and investors who we hoped would participate in the IPO stock offering. Fortunately, our major venture capitalists helped with introductions and advice to guide me through the process. I was also fortunate to have two really bright analysts with the investment banking firms who had been selected to underwrite the IPO offering, and they now understood the science behind structure-based drug design and PNP inhibition. This was critically important because most potential investors relied heavily upon analyst recommendations, especially in emerging fields like biotechnology. However, in early 1994, the IPO markets were beginning to rapidly cool, and there was real urgency to complete our IPO before the funding window completely closed. I was beginning to have nightmares about my decision to abandon my UAB position for a future that would be really tough if we did not complete our IPO. In addition to expected hurdles in putting together the IPO, I was faced with two unusual stumbling blocks. As soon as I moved to BioCryst, I learned that the company was facing a lawsuit from an investment advisor who introduced our two lead venture capitalists to BioCryst. He had expected to receive a commission from this transaction, but it turned out that he was not qualified to conduct security transactions in Alabama, and the Alabama Securities Commission instructed BioCryst that they were not allowed to pay any fee to that agent. He was unqualified because he was a convicted felon in Florida, who had served prison time for burglarizing homes and then burning down the houses to cover the crimes. This agent was determined to force us into payment by doing everything possible to threaten our public stock offering. He repeatedly placed threatening phone calls to our underwriters, analysts, and accounting firm, warning that BioCryst was operating under a lawsuit that threatened to destroy the long term viability of the company. It became a running joke among my New York contacts that they needed to expand their fire insurance coverage if they were going to deal with BioCryst. On top of this major distraction, I was faced with our national accounting firm auditor who was somewhat inexperienced and shocked to learn that BioCryst was not profitable and was not likely to be profitable in the near future. Of course, essentially none of the other biotechnology companies completing IPOs during that period could meet his criteria, but he held up our process for weeks while he conducted a detailed audit of our finances. Despite all of these frustrations, and many sleepless nights for me, we successfully completed our IPO on March 7, 1994 and initiated trading on the NASDAQ stock exchange under the stock symbol BCRX. At the traditional closing dinner in New York, I was presented a fire extinguisher and a smoke detector and wished good luck running a publicly traded company. Then I started receiving a report card every day when the closing price of our stock was posted. I had not received any report cards since I had been promoted to Professor at UAB, except for pink sheets from grant reviews, and now I was being graded daily. I quickly learned that investor relations would be an ongoing part of my new life and that everything I did or did not do was under the close supervision of the Securities and Exchange Commission. Immediately after completing the IPO, I began my formal education on clinical trials. The previous CEO was kind enough to remain long enough to help me understand what had been planned for clinical trials of BCX-34, but my ignorance of the processes involved with clinical protocols, management of trials and clinical data, and filings with the FDA was somewhat overwhelming. We had already embarked on small clinical studies in patients with psoriasis, a T-cell mediated disease that might benefit from PNP inhibition, and we had plans in place to initiate clinical studies of BCX-34 for treatment of patients with cutaneous T-cell lymphoma. Both of these clinical programs were to be conducted initially using a topical formulation of BCX-34, which appeared to have minimal risks to patients. However, I was really concerned about what we were undertaking, largely because I knew so little about clinical development or the ability of our still small company to manage the process. I also did not know our key personnel well enough at that stage to evaluate how effectively they could properly manage the process, even though their resumes appeared impressive. John Montgomery knew more than I did about clinical development since a number of his drugs had advanced through clinical trials while he was at SRI, but he had not been involved with management of those trials. Except for Babu, essentially everyone else in the company was a stranger to me at that stage. I eventually turned to my good friend Nag Nagabhushan at Schering Plough to help me sort through what we should be doing to develop BCX-34 properly. Nag had extensive experience with clinical development programs at Schering Plough, and I knew that I could totally trust his advice. He kindly agreed to come to Birmingham, to review our plans and to interview the key BioCryst personnel who would be responsible for the clinical development program. Nag's analysis after a couple of days at BioCryst was somewhat disconcerting. He was not overly impressed by the experience and backgrounds of the personnel who would be responsible for our studies, and he emphasized to me the importance of carefully monitoring every step of the clinical program. At that stage, small phase 2 trials in patients were already underway, with initial results that had been reported as encouraging. There were no apparent safety issues, which would be of utmost concern to us in moving forward. After Nag's input, I felt that I needed to get someone at BioCryst who had the clinical credentials to oversee our drug development programs. I especially wanted someone that I knew well and could trust completely. My thoughts quickly turned to Bill Cook, my previous graduate student and colleague at UAB. Bill had obtained his PhD degree with me through the UAB MD/PhD program. After completing medical school and a residency in pathology, he had combined a stellar career in clinical pathology at UAB with his continued research in crystallography. He had not been involved directly in clinical trials, but I felt that he had the background and intelligence necessary to quickly understand the clinical development program at BioCryst. First and foremost, however, I knew Bill extremely well and had complete confidence that I could trust his judgment and integrity. I was delighted and relieved when Bill agreed to move to BioCryst. He took over as Senior Vice President of Research and assumed the important position of Medical Director, responsible for overseeing our clinical development programs. Bill was a wonderful addition to BioCryst since he was able to relate to essentially all aspects of our research and development programs and support Babu's ongoing leadership in drug design. He had an impressive background in crystallography, and he had been involved in all aspects of the PNP program since its inception. He also had a sound clinical background and was able to interact effectively with all of our researchers who were responsible for the clinical trials. He immediately added to the credibility of our research activities, and he was a great help to me in interfacing with analysts and investors who were constantly evaluating our programs. Soon after joining BioCryst, Bill undertook a detailed analysis of our clinical data from the initial small phase 2 studies of topical BCX-34 for treatment of skin lesions in patients with psoriasis and patients with cutaneous T-cell lymphoma. Results from these placebo-controlled studies had been publicly announced as positive, and Bill was in the process of working with our head of clinical development to prepare a publication on the detailed results. After working through a weekend with the data, Bill called me on a Sunday afternoon and informed me that he was unable to reproduce the positive results that had been reported. Working with the primary data from the clinical sites, Bill calculated that there was actually no significant difference between the placebo and the drug treated groups. He concluded that altered data had been transferred to the statisticians for analysis, and he saw no way that this could be an honest mistake. Our head of clinical development was confronted with these findings, and he eventually admitted that he had modified the randomization tables in order to make the negative results appear to be strongly positive. We immediately fired him, and early the next morning, we faxed a letter to the FDA notifying them what we had discovered. We later held a telephone conference call open to the investment community to explain, as best we could, what had happened and what we planned to do next in the clinical development program. Falsification of clinical data is a felony, and it would have been a disaster for BioCryst if Bill had not discovered this fabrication of data at an early stage. The FDA sent a team into BioCryst to go through all of our records to completely understand what had occurred and what personnel were involved in the data falsification. They then turned the case over to the Justice Department, which led to an indictment and trial of our ex-head of clinical development, resulting in his successful prosecution. After detailed investigation by the Justice Department, it was concluded that only this one person was involved at BioCryst but that he had colluded with his wife, who worked at one of the clinical trial sites, to fabricate positive clinical results that would allow them to sell BioCryst stock at a profit. They were both convicted and sentenced to terms in prison. Meanwhile, we went on to run larger clinical trials of BCX-34 in patients with psoriasis and cutaneous T-Cell lymphoma, under management of a prominent Clinical Research Organization (CRO). We eventually concluded that BCX-34 was not a suitable candidate for further clinical development. Although it is a potent inhibitor of PNP, the clinical data suggested that the pharmacokinetics of the compound were not adequate for further clinical development. Warner Lambert Pharmaceutical Company completed Phase-1 clinical trials with BCX-5 that indicated this second compound was also unsuitable for further clinical development. Consequently, we renewed our search for a PNP inhibitor that would have better biological properties. Meanwhile, Vern Schramm and his colleagues at the Albert Einstein College of Medicine (AECOM) had designed more potent PNP inhibitors by retaining the heterocyclic ring system of BCX-34 and BCX-5 and replacing the substituent on the 9-position of the heterocyclic ring with various positively charged, nitrogen-containing side chains that formed strong contacts in the sugar-binding site of the enzyme. These compounds seemed to have greatly improved pharmacokinetic properties compared to BCX-34 and BCX-5, so BioCryst entered into a license agreement with AECOM for rights to develop these compounds. Two of these compounds entered advanced stages of clinical development. One of these, BCX-1777 (generic name forodesine, Fig. 26), was eventually licensed to the UK based pharmaceutical company Mundipharma for development in oncology. A second PNP inhibitor, BCX-4208 (generic name ulodesine, Fig. 26), was licensed for a while to Roche for the treatment of psoriasis, but Roche eventually returned the rights to BioCryst. BioCryst continued development through Phase 2 clinical trials for treatment of gout.
Figure 26: Some of the BioCryst compounds that reached advanced stages of development. An especially frustrating design program was our multi-year effort to develop clinically useful inhibitors of the viral enzyme, RNA polymerase. This enzyme is critical for all RNA viruses, including influenza, hepatitis C, and a host of really bad viruses such as Ebola and Marburg. Since the enzyme tends to be reasonably conserved among these viruses, inhibitors of the polymerase might be successful against multiple viral targets. We spent several frustrating years focusing on RNA polymerase from hepatitis C. We successfully designed several potent inhibitors of the enzyme, but our nucleoside analog clinical candidates all proved to have preclinical toxicity profiles that were challenging for clinical development. More recent efforts have resulted in the discovery by BioCryst that a compound in the portfolio of molecules licensed from AECOM is a potent inhibitor against hemorrhagic filoviruses, including Marburg and Ebola [124]. This compound (BCX-4430, Fig. 26) is currently under active development by BioCryst for treatment of Marburg and Ebola viral infections, with funding from the NIAD division of the National Institutes of Health. NIAD recently awarded BioCryst a contract to develop BCX-4430 through Phase 1 for treatment of Ebola virus disease. A dose ranging study of BCX-4430 in nonhuman primates infected with Ebola demonstrated an antiviral effect and showed statistically significant survival. BCX-4430 is now in a Phase 1 clinical trial. Bill Cook gave BioCryst two very productive years before returning as Professor in the Department of Pathology at UAB and Investigator in the Center for Macromolecular Crystallography (since renamed The Center for Structural Biology). We again turned to UAB for leadership at BioCryst and were fortunate to recruit George Omura, a prominent Hematologist/Oncologist in the UAB Comprehensive Cancer Center, to serve as our Medical Director. At the time, George was also a Member of the FDA's Oncology Division Advisory Committee (ODAC), which was responsible for reviewing applications for approval of new oncology drugs, and he had considerable experience with the design of clinical trials in oncology. This was important for BioCryst, since our PNP inhibitor program was focused primarily on the potential of these drug candidates for treatment of T-cell cancers. In 1996, we were also fortunate to recruit Dr. J. Claude Bennett from UAB to serve as our President and Chief Operating Officer. Claude had been a member of our Scientific Advisory Board since its inception, so he was quite familiar with our programs. He was a wonderful addition to BioCryst, since he was a prominent basic scientist and clinician, with extensive administrative experience. He previously served as Chairman of the Department of Microbiology, Chairman of the Department of Medicine, and Founder and Director of the Center For Arthritis Research at UAB. Immediately prior to joining BioCryst, Claude also served several years as President of UAB. I met with him the day he announced his retirement from the Presidency, and I was delighted when he quickly agreed to join BioCryst. Claude is a talented physician and researcher, and he is widely recognized in the medical community. He was on the faculty at UAB when I joined in 1968, so I knew him extremely well and long before we formed BioCryst. He sits through the chemistry and drug design meetings at BioCryst, and he understands what we are doing, while also overseeing our clinical development programs. Along with Babu, Claude completed a science and clinical leadership team that I felt was among the best in the biotech industry. Under Babu's supervision, the drug design group had impressive success with the development of inhibitors of influenza neuraminidase and serine proteases. The PNP and neuraminidase projects proved to be wonderful learning experiences for guiding future design work, since both enzymes crystallized with packing schemes that permitted ready access to their active sites by diffusion of compounds through the solvent channels in preformed crystals. Consequently, it was possible to perform iterative design of potent inhibitors of these two targets by modeling potential compounds using the native structure, binding the compounds directly to the active site by diffusion into native enzyme crystals, determining the structure of the complex, and seeing directly what additional changes to the inhibitor might be likely to further enhance binding. The PNP project ended up determining the crystal structures of approximately forty complexes that were examined through this iterative process and yielded a wealth of information about factors that would be useful in future design projects. This approach of iterative design also proved to be helpful in making structural changes to improve the clinical potential of potent inhibitors that had undesirable properties, such as toxicity, low solubility, poor bioavailability, poor pharmacokinetics or metabolic instability. By seeing directly what parts of an inhibitor might be modified, without altering the binding interactions, it was often possible to work around problems that prevented a good inhibitor from being a suitable drug candidate. Although the original PNP structural studies were performed using synchrotron radiation, the structures of PNP complexes at BioCryst were determined using our laboratory based area detector system, which was equipped with a rotating anode generator, low temperature system and focusing optics. All of the neuraminidase work, which eventually involved determining the structures of some 120 inhibitor complexes during design optimization, also was done using data collected with our in-house facilities. Thus, we could get fairly rapid turnaround of structural information by designing changes to a lead compound, synthesizing the new compound in house, immediately seeing exactly how it binds to the target in the crystal structure of the complex, and modeling reasonable additional changes to enhance the properties of the inhibitor. Following this iterative approach, Babu's team developed peramivir (Fig. 26), a potent inhibitor of influenza neuraminidase. We entered into a partnership with Johnson and Johnson (J&J) for worldwide clinical development of peramivir in 1999. J&J advanced peramivir up through early Phase 3 U.S. and international clinical trials before deciding that low oral bioavailability of the compound was unsuitable for their commercialization goals. We had made extensive efforts to improve the oral bioavailability of peramivir, without success. The compound had wonderful activity as an intravenous agent, but J&J's commercial interest was restricted to an oral drug. Consequently, J&J returned all rights for peramivir to BioCryst, including an extensive package of clinical and preclinical data and a process for manufacturing the compound. The clinical studies had demonstrated a good safety profile for peramivir, and later in vitro tests against new emerging strains of influenza demonstrated that the compound has activity against multiple strains of influenza, including avian strains that have been of increasing concern as possible pandemic threats. BioCryst turned its focus to development of an intravenous formulation of peramivir. Governments around the world became increasingly concerned about the threat of emerging deadly strains of influenza, and funds were appropriated through HHS (Health and Human Services, the parent of NIH) to support new therapies for treatment of influenza. We had a strong head start with the development of intravenous peramivir, including a feasible manufacturing process, and we were successful in our application for support from HHS for development of the intravenous drug. (BioCryst has now received over $200 million from HHS and the Biomedical Advanced Research and Development Authority, BARDA, for the development of peramivir.) At about the same time, we were approached by Shionogi, a Japanese pharmaceutical company, for the rights to develop peramivir in Japan. In Japan, it is not unusual for patients with fairly routine illnesses to be treated by intravenous formulations in doctors' offices, and Shionogi saw the potential of using peramivir for the treatment of seasonal flu, in addition to any potential pandemic applications. Shionogi successfully completed clinical trials in Japan, which demonstrated that a single intravenous infusion of peramivir is effective for treating seasonal influenza. The intravenous drug is now on the market in Japan, under the trade name of Rapiacta. Peramivir is also approved in South Korea, by Green Cross Pharmaceuticals, under the trade name Peramiflu. Meanwhile, BioCryst conducted additional clinical trials with intravenous peramivir (trade name Rapivab) through HHS/BARDA funding. In December 2014 the FDA approved Rapivab (peramivir injection) as a single-injection treatment of uncomplicated influenza in adults. This was the first new antiviral treatment for influenza approved by the FDA in 15 years. It was also the first BioCryst designed drug to be approved by the FDA for marketing in the U.S. The BioCryst program directed at serine protease inhibitors has been ongoing ever since the IPO in 1994. Our initial focus was on inhibitors of the complement enzyme Factor D, in collaboration with investigators from UAB [163, 167]. We successfully designed a potent Factor D inhibitor (BCX-1470, Fig. 26) that entered Phase 1 trials, but the trials were discontinued due to a less than ideal profile of the compound. The next target that was approached in this series was the tissue factor-factor 7A complex, a target for cardiovascular applications. This program resulted in BCX-4161, a potent inhibitor of plasma kallikrein, another serine protease. Kallikrein is the culprit in the disease hereditary angioedema (HAE), a devastating chronic ailment that impacts the lives of several thousand patients in the U.S. Injectable formulations of kallikrein inhibitors are on the market, but these require frequent infusions. BCX-4161 is orally active, meaning that patients might be treated by taking daily pills, which would be a significant improvement in their quality of life. BCX-4161 has completed a successful phase 2 clinical trial in patients with HAE, and is currently in a larger Phase 2 trial on the path to potential approval for marketing the drug in the U.S. Meanwhile, Babu and his team are making good progress with the design of improved, second generation inhibitors of kallikrein. At this stage, they have the experience that allows successful design working only with the native structure of the enzyme, without necessarily resorting to iterative crystallographic analyses of multiple complexes. Over the years, they have become remarkably proficient at using the extensive experience they gained with the PNP and neuraminidase programs for rapidly designing effective inhibitors of other targets. The key scientists in the medicinal chemistry group have now been working with Babu for a number of years, and the speed and efficiency with which this group is able to go from the structure of a protein target to the design of effective inhibitors is amazing. It is still a challenge to design compounds that can pass successfully through the torturous path of successful clinical development, but I feel that our original goal of seeing structure-based drug design developed into a powerful tool at BioCryst has been achieved. In 2006, I announced my intent to retire as CEO of BioCryst in 2007, the year I would reach age 65. The company had reached the stage where the focus needed to be on final approval of our drug candidates and commercialization of these drugs. It seemed likely that we could now recruit an experienced CEO from the pharmaceutical industry who had the experience and credentials to move our drug pipeline through final development and to market. We had established a BioCryst division in 2006 at the Research Triangle in North Carolina to oversee our clinical development and regulatory (i.e., FDA related) activities. That area in North Carolina is one of the best places in the world to manage clinical development due to the heavy concentration of contract research organizations and pharmaceutical companies. The headquarters for BioCryst were moved to North Carolina, after the company recruited Jon Stonehouse to replace me as CEO of BioCryst. All of the research functions have remained in Birmingham under the leadership of Babu who is doing a superb job continuing the structure-based design program. I stayed on as Chairman of the Board until our annual meeting in May 2007. I now maintain an office at the company in Birmingham, and BioCryst has been generous to keep me on as a consultant. Claude Bennett retired a couple of years after I did, and he and I meet weekly with the drug design team. I am looking forward to finally seeing the drugs that we have worked on all these years reaching patients around the world. Given the progress the company has made recently, I am optimistic that we are getting close to that goal. So what have I learned through these years in the biotechnology industry? First and foremost, it is incredibly difficult and expensive to develop a drug, and the risks involved in moving a compound successfully through the development process are immense. The FDA typically approves 20-30 new drugs each year, although they have done a little better than that recently. The last financial figures I saw, which are probably now under estimates, indicate that the pharmaceutical and biotechnology industries spend over $50 billion dollars a year on research and development, so it is easy to see how expensive it is, on the average, to produce that handful of drugs. A very recent analysis from Tufts University concluded that the average cost of developing a drug currently exceeds $2 billion. What is the chance of a given compound making it successfully through the development process? I have seen figures ranging from 1/500 to 1/10,000. Our experience at BioCryst indicates that those odds are improved by systematic use of structural data during the design and drug optimization process, but a number of initially promising compounds still fail during the clinical stage of development. How long does it take to get a drug from discovery to patients? We started BioCryst in 1986, building initially on several years of research already completed at UAB and SRI, so our experience certainly suggests that it can take many years to get drugs successfully through the development process. The BioCryst drug development programs have required extensive funding over the years, which has come from periodic stock sales, government grants and contracts, and support from our pharmaceutical partners; but we have still spent considerably less than the average cost involved in getting drugs to market. Maybe that is attributable to the added efficiency of structure-based design, but we will have to wait and see when the BioCryst compounds now in development reach the market. Above all else, it is clear to me that structure-based design allows a small, focused team to undertake pharmaceutical design and development projects that have generally been the sole purview of large pharmaceutical companies. The economics of a drug discovery and development company like BioCryst are interesting and somewhat unique. BioCryst has operated in the red, meaning without profits, ever since our founding in 1986. This is not completely surprising considering the long time generally required to move a drug successfully from design, through clinical development and through FDA approval processes. Despite this, BioCryst has remained solvent ever since completing our IPO in 1994. There have been periods when we had to cut back programs and decrease personnel, but we were never really concerned about our financial survival. We generally strived to maintain a bank balance that would support the company for at least a year, and we were able to do this through periodic public and private sales of stock. Many of the development costs of the drug candidates have been funded by pharmaceutical partners, and BioCryst has also benefitted from substantial government contracts for developing peramivir and BCX-4430. The deficit between the revenues obtained from these sources and the research and development expenses has been filled over the years by multiple equity offerings. The ability to raise this capital in the equity markets is highly dependent on BioCryst's status as a publicly traded company, which was the original carrot that lured me from academia to pursue the dream of using crystallography to develop important drugs that might eventually make a big difference in the lives of patients.
Figure 27: BioCryst opening the NASDAQ stock exchange on the morning of September 8, 2006, in celebration of our twentieth year anniversary. Shown from left to right (beyond the half head view of a NASDAQ official) are my longtime Administrative Assistant, Penny Mann; Bebe; me; Mike Darwin, the BioCryst CFO; a NASDAQ official; and a partial head view of Randy Riggs, BioCryst Vice President for Commercial Development. Before I actually retired as CEO, I was invited to open trading (ring the opening bell) at the NASDAQ stock exchange in recognition of BioCryst's twenty-year anniversary (Fig. 27). Bebe and several colleagues from the company, including my longtime Administrative Assistant, Penny Mann, joined me. I gave a short talk on the history of the company, which presumably is filed away somewhere in the archives of the stock exchange. However, the main highlight was the picture of me and Bebe together, which was shown off and on during the day on the Jumbotron at Times Square (Fig. 28). I have a blown-up copy of this picture framed in my bathroom at home to remind me each morning of the many exciting, fun and stimulating paths crystallography has allowed me to follow and enjoy during my career. I can't even begin to count all of the lucky breaks in my life that led me into a career in crystallography, and I cannot imagine any field that could have been more rewarding. It is mind boggling to realize the large number of Nobel Laureates and other brilliant colleagues I have known personally and have had the chance to work with during my career. What other field could possibly offer such opportunities? I have also been able to witness and live through major revolutions in applications of crystallography, and I already see exciting new forefronts opening up in biology. My major goal at this stage is to totally enjoy life and to stay alive and healthy for a long time to witness the revolutions that are yet to come. Enjoying life is real easy, with my three children (Jeannie, Betsy and Eddie) and six grandchildren (Brooke, Thomas, Banks, Lauren, Bebe, and John) living only a few blocks away from me and Bebe. My three children all have very productive and successful careers. I have not yet produced a crystallographer out of this group, but my son Eddie is a prominent surgeon. My father is probably happy with that.
Figure 28: Bebe and I featured on the NASDAQ Jumbotron in Times Square. This display was shown periodically during the day, in celebration of BioCryst's anniversary. This picture has been blown up to poster size, and is now mounted on the wall of my bathroom at home.
References 1.Bugg, C.E., Lawson, J. and Sass, R.L. The Crystal Symmetry of Several Diazonium Salts. Acta Cryst. 17: 767-768 (1964). 2.Bugg, C.E., Desiderato, R. and Sass, R.L. An X-ray Diffraction Study of Nonplanar Carbanion Structures. J. Am. Chem. Soc. 86: 3157 (1964). 3.Bugg, C.E. and Sass, R.L. The Crystal Structure of Pyridinium Dicyanomethylide. Acta Cryst. 18: 591-594 (1965). 4.Sass, R.L. and Bugg, C.E. The Crystal Structure of Potassium Paranitrophenyl-dicyanomethide. Acta Cryst. 23: 282-288 (1967). 5.Bugg, C.E. and Marsh, R.E. Structure of the Monoclinic Form of Cytidylic Acid b. J. Mol. Biol. 25: 67-82 (1967). 6.Bugg, C.E., Thewalt, U. and Marsh, R.E. Base Stacking in Nucleic Acid Components: The Crystal Structure of Guanine, Guanosine and Inosine. Biochem. Biophys. Res. Comm. 33: 436-440 (1968). 7.Bugg, C.E. and Thewalt, J. Effects of Halogen Substituents on Base Stacking in Nucleic Acid Components: The Crystal Structure of 8-Bromoguanosine. Biochem. Biophys. Res. Comm . 37: 623-629 (1969). 8.Thewalt, U., Bugg, C.E. and Hettche, A.Structure of1,1-Bis(dimethylamino)-2,4,6- triphenylphosphorinanium. Angew. Chem. Int. Ed. 9: 898-899 (1970).
9.Bugg, C.E., Thomas, J.M., Sundaralingam, M. and Rao, S.T. Stereochemistry of Nucleic Acids and Their Constituents. X. Solid-State Base-Stacking Patterns in Nucleic Acid Constituents and Polynucleotides. Biopolymers 10: 175-219 (1971). 10.Bugg, C.E . and Thewalt, U. The Crystal and Molecular Structure of 6-Thioguanine. J. Am. Chem. Soc. 92: 7441-7445 (1970). 11.Subramanian, E., Trotter, J. and Bugg, C.E. Crystal Structure of Ethidium Bromide. J. Cryst. Mol. Struct. 1: 3-15 (1971). 12.Bugg, C.E. and Thewalt, U. Crystal Structure of Serotonin Picrate, A Donor-Acceptor Complex. Science 170: 852-854 (1970). 13.Thewalt, U., Bugg, C.E. and Marsh, R.E. The Crystal Structure of Guanosine Dihydrate and Inosine Dihydrate. Acta Cryst. B26: 1089-1101 (1970). 14.Thewalt, U., Bugg, C.E. and Marsh, R.E. The Crystal Structure of Guanine Monohydrate. Acta Cryst. B27: 2358-2363 (1971). 15.Thewalt, U. and Bugg, C.E. The Crystal and Molecular Structure of Serotonin Picrate Monohydrate. Acta Cryst. B28: 82-92 (1972). 16.Bugg, C.E. and Thewalt, U. Crystal Structure of N6-(δ2-isopentenyl)adenine, A Base in the Anticodon Loop of Some tRNA's. Biochem. Biophys. Res. Comm. 46: 779-784 (1972). 17.Bugg, C.E. and Cook, W.J. Calcium Ion Binding to Uncharged Sugars: Crystal Structure of Calcium Bromide Complexes of Lactose, Galactose and Inositol. J. Chem. Soc., Chem. Comm. 12: 727-729 (1972). 18.Thewalt, U. and Bugg, C.E. The Crystal and Molecular Structure of 1,1-bis(dimethylamino)-2,4,6- triphenylphosphorin. Acta Cryst. B28: 871-879 (1972). 19.Bugg, C.E . Solid State Stacking Patterns of Purine Bases. In: Jerusalem Symposia on Quantum Chemistry and Biochemistry. Vol. IV. The Purines-Theory and Experiment (ed . E.d. Bergmann and B. Pullman), Academic Press, pp. 178-204 (1972). 20.Bugg, C.E. and Thewalt, U. The Crystal Structure of 6-Histamino Purine Dihydrate. Acta Cryst. B28: 1767-1773 (1972). 21.Kolata,G. The Great Crystal Caper. Science 229:370-371(1985). 22.Freeman, G.R., Hearn, R.A. and Bugg, C.E. The Crystal Structure of Glycylglycine Phosphate Monohydrate. Acta Cryst. B28: 2906-2912 (1972). 23.Hearn, R.A. and Bugg, C.E. The Crystal Structure of (-)-Ephedrine Dihydrogen Phosphate. Acta Cryst. B28: 3662-3667 (1972). 24.Thewalt, U. and Bugg, C.E. Effects of Sulfur Substituents on Hydrogen Bonding and Base Stacking: Crystal Structure of 6-Thioguanosine Monohydrate. J. Am. Chem. Soc. 94: 8892-8898 (1972). 25.Bugg, C.E. Calcium Binding to Carbohydrates: Crystal Structure of a Hydrated Calcium Bromide Complex of Lactose. J. Am. Chem. Soc. 95: 908-913 (1973). 26.Subramanian, E., Madden, J.J. and Bugg, C.E . A syn Conformation of Inosine, the Wobble Nucleoside in Some tRNA's. Biochem. Biophys. Res. Commun. 50: 691-696 (1973). 27.Thewalt, U. and Bugg, C.E. Crystal Structure of Calcium Chloride Tetrahydrate. Acta Cryst. B29: 615-617 (1973). 28.Cook, W.J. and Bugg, C.E. Lactose-Calcium Chloride Heptahydrate Complex. Acta Cryst. B29: 907- 909 (1973). 29.Cook, W.J. and Bugg, C.E. Calcium Binding to Carbohydrates: Crystal Structure of a Hydrated Calcium Bromide Salt of Lactobionic Acid. Acta Cryst. B29: 215-222 (1973). 30.Sternglanz, H. and Bugg, C.E. Conformations of N6-Monosubstituted Adenine Derivatives: Crystal Structure of N6-Methyladenine. Biochim. Biophys. Acta 308: 1-8 (1973). 31.Sternglanz, H. and Bugg, C.E. A Cyclitol Pentaacetate (±)2-Acetoxymethyl-1,3,4,6-Tetra-O-Acetyl- Epi-Inositol. Acta Cryst. B29: 1536-1538 (1973). 32.Thewalt, U. and Bugg, C.E. The Crystal Structure of 5-Hydroxyuridine. Acta Cryst. B29: 1393-1398 (1973). 33.Cook, W.J. and Bugg, C.E. Effects of Calcium Interactions on the Conformations of Sugars: Crystal Structure of Myo-Inositol Calcium Bromide Pentahydrate. Acta Cryst. B29: 2404-2411 (1973). 34.Cook, W.J. and Bugg, C.E. Calcium Binding to Galactose: Crystal Structure of a Hydrated alpha- Galactose Calcium Bromide Complex. J. Am. Chem. Soc. 95: 6442-6446 (1973). 35.Sternglanz, H. and Bugg, C.E. Crystal Structure and Chemical Configuration of 2,4-Diamino-5- methyl-6-benzylpyrido 2,3-d pyrimidine hydrobromide. Acta Cryst. B29: 2191-2194 (1973). 36.Hearn, R.E., Freeman, G.R and Bugg, C.E. Conformational and Phosphate Binding Properties of Phenylethanolamines: Crystal Structure of Ephedrine Monohydrogen Phosphate Monohydrate. J. Am. Chem. Soc. 95: 7150-7154 (1973). 37.Sternglanz, H. and Bugg, C.E. Conformation of N6-Methyladenine, A Base Involved in DNA Modification-Restriction Processes. Science 182: 833-834 (1973). 38.Cook, W.J. and Bugg, C.E. Calcium Interactions with D-Glucans: Crystal Structure of alpha, alpha- Trehalose-Calcium Bromide Monohydrate. Carbohyd. Res. 31: 265-275 (1973) 39.Cantrell, C.E., Kiely, D.E., Hearn, R.A. and Bugg, C.E. The Chemistry of Delta Dicarbonyl Sugars. The Bases Catalyzed Cyclization of a Xylo-hepto -2,6-diulose to an Unusual Cyclohexenone. Tetrahedron Letters 44: 4379-4382 (1973). 40.Bugg, C.E. Crystal Chemical Considerations. J. Dental Res. 53: 293-296 (1973). 41.Einspahr, H. and Bugg, C.E. Calcium Binding to alpha-Amino Acids: Crystal Structure of Calcium L-Glutamate Trihydrate. Acta Cryst. B30: 1037-1043 (1973). 42.Freeman, G.R and Bugg, C.E. Geometries of Donor-Acceptor Complexes: Crystal Structure of the Iodide, Chloride and Picrate Salts of 1-Methylnicotinamide. Acta Cryst. B30: 431-443 (1973). 43.Gartland, G.L., Freeman, G.R and Bugg, C.E. Crystal Structures of Tryptamine Picrate and D,L- Tryptophan Picrate Methanol, Two Indole Donor-Acceptor Complexes. Acta Cryst. B30: 1841-1849 (1974). 44.Hearn, R.A. and Bugg, C.E. Calcium Binding to Carbohydrates: Crystal Structure of Calcium Ascorbate Dihydrate. Acta Cryst. B30: 2705-2711 (1974). 45.Sternglanz, H ., Einspahr, H.M. and Bugg, C.E. Structure and Conformation of 4-Peroxycyclophosphamide. Cytotoxic Oxidation Product of Cyclophosphamide. J. Am. Chem. Soc. 96: 4014-4015 (1974). 46.Sternglanz, H . and Bugg, C.E. Relationship Between the Mutagenic and Base-Stacking Properties of Halogenated Uracil Derivatives. The Crystal Structures of 5-Chloro- and 5-Bromouracil. Biochim. Biophys. Acta. 378: 1-11 (1975). 47.Bugg, C.E. and Thewalt, U. Effects of Sulfur Substituents on the Structural Properties of Purines: Crystal Structures of Guanine Picrate Monohydrate and 6-Thioguanine Picrate Monohydrate. Acta Cryst. B31: 121-127 (1975). 48.Freeman, G.R and Bugg, C.E. The Crystal Structure of Procaine Dihydrogen Orthophosphate. Acta Cryst. B31: 96-101 (1975). 49.Bugg, C.E. and Sternglanz, H. Structural Properties of Purine and Pyrimidine Analogs. In: Molecular and Quantum Pharmacology (ed. ED. Bergmann and B. Pullman), Academic Press, Jerusalem, 473-500 (1975). 50.DeLucas, L.J. and Bugg, C.E. Calcium Binding to D-Glucuronate Residues: Crystal Structures of a Hydrated Calcium Bromide Salt of D-Glucuronic Acid. Carbohydr. Res. 41: 19-29 (1975). 51.Cook, W.J. and Bugg, C.E. Crystal Structures of Azathioprine Dihydrate and 6-Methylmercapto- purine Trihydrate. J. Pharm. Sci. 64: 221-225 (1975). 52.Pitner, T.P., Sternglanz, H ., Bugg, C.E. and Glickson, J.D. Proton Nuclear Magnetic Resonance Study of Hindered Internal Rotation of the Dimethylamino Group of N6, N6-Dimethyladenine Hydrochloride in Aqueous Solution. J. Am. Chem. Soc. 97: 885-888 (1975). 53.Cook, W.J. and Bugg, C.E. Calcium-Carbohydrate Bridges Composed of Uncharged Sugars: Structure of a Hydrated Calcium Bromide Complex of alpha-Fucose. Biochim. Biophys. Acta 389: 428-435 (1975). 54.Sternglanz, H ., Freeman, G.R. and Bugg, C.E. Crystal Structure of 5-Iodouracil. Acta Cryst. B31: 1393-1395 (1975). 55.Sternglanz, H. and Bugg, C.E. Crystal Structure and Base Stacking Pattern of 6-Chloropurine Riboside. Acta Cryst. B31: 2888-2891 (1975). 56.Cook, W.J., Thewalt, U. and Bugg, C.E . Calcium Binding to Non-Ionic Lipids: Crystal Structure of a Geraniol-Calcium Chloride Complex. Biochem. Biophys. Res. Comm. 68: 143-148 (1976). 57.Cook, W.J. and Bugg, C.E. Effects of Calcium Interactions on Sugar Conformation: Crystal Structure of beta-D-Fructose-Calcium Bromide Dihydrate. Acta Cryst. B32: 656-659 (1976). 58.Sternglanz, H ., Subramanian, E., Lacey, J.C. and Bugg, C.E. Interactions of Hydrated Metal Ions with Nucleotides: The Crystal Structure of Barium Adenosine-5'-Monophosphate. Biochem. 15: 4797-4802 (1976). 59.Cook, W.J. and Bugg, C.E. Structures of Calcium-Carbohydrate Complexes. In: Metal-Ligand Interactions in Organic and Biochemistry (ed. B. Pullman and N. Goldblum), D. Reidel Publishing Co., part 2, pp. 231 -256 (1977). 60.Abruscato, G.J., Kiely, D.E., Cook, W.J. and Bugg, C.E. Delta Dicarbonyl Sugars. VI. Preparation of an Unusual Trihaloheptulose from Xylaric Acid. J. Org. Chem. 42: 3567-3571 (1977). 61.Sternglanz, H ., Thomas, J.M. and Bugg, C.E. Stacking Patterns of Halogenated Purines: Crystal Structure of 8-Bromoinosine. Acta Cryst. B33: 2097-2101 (1977). 62.DeLucas, L.J., Hearn, R.A. and Bugg, C.E. Stacking Patterns of Thiopyrimidines: Crystal Structure of 2-Thiocytosine Picrate. Acta Cryst. B33: 2611-2614 (1977). 63.Gartland, G.L. and Bugg, C.E. The Crystal Structure of 2'-Deoxy-6-Thioguanosine Monohydrate. Acta Cryst. B33: 3678-3681 (1977). 64.Einspahr, H.M., Gartland, G.L. and Bugg, C.E. Calcium-Binding to Alpha-Amino Acids: Crystal Structure of Calcium L-Glutamate Chloride Monohydrate. Acta Cryst. B33: 3385-3390 (1977). 65.Einspahr, H.M. and Bugg, C.E. Crystal Structures of Calcium Complexes of Amino Acids, Peptides and Related Model Systems. In: Calcium-Binding Proteins and Calcium Function (ed. RH. Wasserman), North-Holland, pp . 13-20 (1977). 66.Einspahr, H.M., Bugg, C.E. and Cook, W.J. Geometry of Calcium: Water Interactions in Crystalline Hydrates. In: Calcium-Binding Proteins and Calcium Function (ed. RH. Wasserman), North-Holland, pp. 76-77 (1977). 67.Adams, M.J., Helliwell, J.R. and Bugg, C.E. Structure of 6-Phosphogluconate Dehydrogenase from Sheep Liver at 6A Resolution. J. Mol. Biol. 112: 183-197 (1977). 68.DeLucas, L.J., Suddath, F.L., Gams, R.A. and Bugg, C.E. Preliminary X-ray Study of Crystals of Human Transferrin. J. Mol. Biol. 123: 285-286 (1977). 69.Abrahams, S.C., Bernstein, J.L., Bugg, C.E. and Hvoslef, J. Comparison of Two Independent Determinations of the Structure of Calcium L-Ascorbate Dihydrate. Acta Cryst. B34: 2981-2985 (1978). 70.DeLucas, L.J., Gartland, G.L. and Bugg, C.E. Sodium Binding to D-glucuronate Residues: Crystal Structure of Sodium D-glucuronate Monohydrate. Carbohyd. Res. 62 : 213-221 (1978). 71.Fontecilla-Camps, J.C., Bugg, C.E., Temple Jr., C., Rose, J.D., Montgomery, J.A. and Kisliuk, R.L. x-ray Crystallography Studies of the Structure of 5)0-Methenyl-Tetrahydrofolic Acid. In: The Chemistry and Biology of Pteridines (ed. G.M. Brown), Elsevier-North Holland, pp . 235-240 (1978). 72.Fontecilla-Camps, J.C., Suddath, F.L., Bugg, C.E. and Watt, D.D. Crystals of a Toxic Protein from the Venom of the Scorpion Centruroides sculpturatus Ewing: Preparation and Preliminary X-ray Investigation. J. Mol. Biol. 123: 703-705 (1978). 73.Einspahr, H .M. and Bugg, C.E. Calcium Binding to Alpha-Amino Acids: Crystal Structure of Calcium Di-L-Glutamate Tetrahydrate. Acta Cryst. B35: 316-321 (1979). 74.Meehan Jr., E.L., Einspahr, H.M. and Bugg, C.E. Calcium Binding to Hydroxycarboxylic Acids: Crystal Structure of Calcium D, L-Diglycerate Dihydrate. Acta Cryst. B35: 828-832 (1979). 75.Sternglanz, H., Graves, D.E., Yielding, L.W. and Bugg, C.E. Crystal Structure of Ethidium Monoazide, A Photoactive Compound that Reacts with Nucleic Acids. J. Cryst. Mol. Struct. 8: 93-103 (1978). 76.Cook, W.L., Suddath, F.L., Bugg, C.E. and Goldstein, G. Crystallization and Preliminary X-ray Investigation of Ubiquitin, A Nonhistone Chromosomal Protein. J. Mol. Biol. 130: 353-355 (1979). 77.Einspahr, H.M. and Bugg, C.E. Enamel Apatite and Caries - A Crystallographic View. In: The Biologic Basis of Dental Caries, Ch. 7 (ed. L. Menaker), Harper & Row, Hagerstown, MD (1980). 78.Sternglanz, H. and Bugg, C.E. Conformation of N(6)-Substituted Adenine Derivatives: Crystal Structures of N(6)-Methyladenine Hydrochloride and N(6), N(9)-Dimethyladenine. J. Cryst. Mol. Struct. 8: 263-277 (1978). 79.Fontecilla-Camps, J.C., Bugg, C.E., Temple Jr., C., Rose, J.D. and Montgomery, J.A. Absolute Configuration of Biological Tetrahydrofolates: A Crystallographic Determination. J. Am. Chem. Soc. 101: 6114-6115 (1979). 80.DeLucas, L.L. Einspahr, H.M. and Bugg, C.E. Calcium Binding to Amide Carbonyl Groups: Structure of a Calcium Bromide Salt of D-Panthotenic Acid. Acta Cryst. B35: 2724-2726 (1979). 81.Einspahr, H.M. and Bugg, C.E. The Geometry of Calcium-Water Interactions in Crystalline Hydrates. Acta Cryst. B36: 264-271 (1980). 82.Brown, E.A. and Bugg, C.E. Calcium Binding to Nucleotides: Crystal Structure of a Hydrated Calcium Salt of Inosine-5'-Monophosphate. Acta Cryst. B36: 2597-2604 (1980). 83.Cook, W.L Einspahr, H .M., Trapane, T.L., Urry, D.W. and Bugg, C.E. The Crystal Structure and Conformation of the Cyclic Trimer of a Repeat Pentapeptide of Elastin, Cyclo-(L-Valyl-L-Prolyl-Glycyl-L-Valyl-Glycyl) J. Am. Chem. Soc. 102: 5502-5505 (1980). 84.Cook, W.L., Gartland, G.L. and Bugg, C.E. Conformation of an O(6)-Substituted Purine Derivative: Crystal Structure of 6-Methoxypurine Riboside. Acta Cryst. B36: 2467-2470 (1980). 85.Brown, E.A. and Bugg, C.E. Binding of Alkaline Earth Metal Ions to Nucleotides: Crystal Structure of a Hydrated Strontium Salt of Inosine-5'-Monophosphate. J. Cryst. Mol. Struct. 10: 19-30 (1980). 86.Riordan, J.M., Kiely, D.E., DeLucas, L.L., Einspahr, H.M. and Bugg, C.E. Branched-Chain Halo and Aminocyclitols: Synthesis and an X-ray Crystallographic Study. Carbohyd. Res. 82: 303-324 (1980). 87.Cook, W.J., Dedman, J.R., Means, A.R. and Bugg, C.E. Crystallization and Preliminary X-ray Investigation of Calmodulin. J. Biol. Chem. 255: 8152-8153 (1980). 88.Cook, W.J., Bugg, C.E., Dedman, J.R. and Means, A.R Crystallographic Studies of Calmodulin. Annals N. Y. Acad. Sci. 356: 365-366 (1980). 89.Einspahr, H. and Bugg, C.E. The Geometry of Calcium-Carboxylate Interacations in Crystalline Complexes. Acta Cryst. B37: 1044-1052 (1981). 90.Fontecilla-Camps, J.C., Almassy, R.J., Suddath, F.L., Watt, D.D. and Bugg, C.E. Three-Dimensional Structure of a Protein from Scorpion Venom: A New Structural Class of Neurotoxins. Proc. Nat. Acad. Sci. USA 77: 6496-6500 (1980). 91.Cook, W.J., Ealick, S.E., Bugg, C.E., Stoeckler, J.D. and Parks Jr., R.E. Crystallization and Preliminary X-ray Investigation of Human Erythrocytic Purine Nucleoside Phosphorylase. J. Biol. Chem. 256: 4079-4080 (1981). 92.Einspahr, H.M., Cook, W.J. and Bugg, C.E. Conformational Flexibility in Single-Stranded Oligonucleotides: Crystal Structure of a Hydrated Calcium Salt of Adenylyl-(3',5')-Adenosine. Biochem. 20: 5788-5794 (1981). 93.Fontecilla-Camps, J.C., Almassy, R.J., Ealick, S.E., Suddath, F.L., Watt, D.D., Feldmann, R.J. and Bugg, C.E. Architecture of Scorpion Neurotoxins: A Class of Membrane-Binding Proteins. Trends. Biochem. Sci. 6: 291-296 (1981). 94.Suddath, F.L., Ealick, S.E., Almassy, R.J., Fontecilla-Camps, J.C. and Bugg, C.E. The 1.3A Structure of a Scorpion Neurotoxin, a Membrane-Binding Protein. Biophys. Jour. 37: 185-186 (1982). 95.Fontecilla-Camps, J.C., Almassy, R.J., Suddath, F.L. and Bugg, C.E. The Three-Dimensional Structure of a Scorpion Neurotoxin. Toxicon 20: 1-7 (1982) . 96.Meehan Jr., E.J., McDuffie, J., Einspahr, H.M., Bugg, C.E. and Suddath, F.L. The Crystal Structure of Pea Lectin at 6.0A Resolution. J. Biol. Chem. 257: 13278-13282 (1982). 97.Hogan, A, Ealick, S.E., Bugg, C.E. and Barnes, S. Aggregation Patterns of Bile Salts: Crystal Structure of Calcium Cholate Chloride Heptahydrate. J. Lipid Res . 25: 791-798 (1984). 98.Almassy, R.J., Fontecilla-Camps, J.C., Suddath, F.L. and Bugg, C.E. Structure of Variant-3 Scorpion Neurotoxin from Centruroides sculpturatus Ewing Refined at 1.8A. J. Mol. Biol. 170: 497-527 (1983). 99.Einspahr, H.M. and Bugg, C.E. Crystal Structure Studies of Calcium Complexes and Implications for Biological Systems. In: Metal Ions in Biological Systems, Vol. 17: Calcium and Its Role in Biology (ed. H. Sigel), Marcel Dekker: Basel, Switzerland, pp. 51-97 (1983). 100.Krishna, N.R, Bugg, C.E., Stephens, R.L. and Watt, D.D. NMR Studies of the Variant-3 Neurotoxin from Centruroides sculpturatus Ewing. J. Biomolec. Struct. and Dynamics 1: 829-842 (1983). 101.Adams, M.J., Archibald, I.G., Bugg, C.E., Carne, A., Gover, S., Helliwell, J.R., Pickersgill, R.W. and White, S.W. The Three-Dimensional Structure of Sheep Liver 6-Phosphogluconate Dehydrogenase at 2.6A Resolution. Embo. J. 2: 1009-1014 (1983). 102.Sack, J.S., Bugg, C.E. and Cook, W.J. Crystal Structure of Calmodulin. In: Calcium-Binding Proteins in Health and Disease (ed. E. Carafoli et al.), pp. 163-164 (1983). 103.Ealick, S.E., Cook, W.J., Fontecilla-Camps, J.C, Suddath, F.L., Bugg, C.E. and Watt, D.D. Preliminary X-ray Investigation of Variant-2 Scorpion Toxin from Centruroides sculpturatus Ewing: Evidence of a Reversible Transition Between Crystal Forms. J. Biol. Chem. 259: 12081-12083 (1984). 104.Zell, A., Einspahr, H .M. and Bugg, C.E. Calcium Binding to Gamma-Carboxyglutamic Acid Residues of Proteins. Crystal Structure of Calcium Alpha-Ethylmalonate. Biochem. 24: 533-537 (1984). 105.Smith, C.D., DeLucas, L.J., Ealick, S.E., Schweitz, H ., Bugg, C.E. and Lazdunski, M. Crystallization and Preliminary X-ray Investigation of a Protein Neurotoxin from the Sea Anemone Anthopleura xanthograrnrnica. J. Biol. Chem. 259: 8010-8011 (1984). 106.Zell, A., Ealick, S.E. and Bugg, C.E. Three-Dimensional Structures of Scorpion Neurotoxins. In: Molecular Architecture of Proteins and Enzymes, II U.S. China Conference on Proteins (ed . Bradshaw, R. and Tang, J.), Academic Press, NY, pp. 65-97 (1985). 107.Vijay-Kumar, S., Bugg, C.E., Wilkinson, K.D. and Cook, W.J. Three-Dimensional Structure of Ubiquitin at 2.8A Resolution. Proc. Nat. Acad. Sci. USA 82: 3582-3585 (1985). 108.Babu, Y.S., Sack, J.S., Greenhough, T.G., Bugg, C.E., Means, A.R. and Cook, W.J. Three-Dimensional Structure of Calmodulin. Nature 315: 37-40 (1985). 109.Cook, W.J., Ealick, S.E., Krenitsky, T.A., Stoeckler, J.D., Helliwell, J.R. and Bugg, C.E . Crystallization and Preliminary X-ray Investigation of Purine Nucleoside Phosphorylase from Escherichia coli. J.Biol.Chem. 260: 12968-12969 (1985). 110.Ealick, S.E., Greenhough, T.G., Babu, Y.S., Carter, D.C., Cook, W.J., Bugg, C.E., Rule, S., Habash, J., Helliwell, J.R., Stoeckler, JD., Chen, S. and Parks Jr., R. Three-Dimensional Structure of Human Erythrocytic Purine Nucleoside Phosphorylase. Annals N.Y. Acad. Sci. 451: 311-312 (1986). 111.Nettesheim, D.G., Bugg, C.E., Krishna, N.R. and Watt, D.D. A Preliminary Study of the Ring- Current Induced Shifts in the NMR Spectrum of the Variant-3 Neurotoxin from Centruroides sculpturatus Ewing. In : Peptides: Structure and Function (ed. Kobble, K. et al.), Pierce Chemical Co., Rockford, IL, pp. 843-846 (1985). 112.Stoeckler, J.D., Ealick, S.E., Bugg, C.E. and Parks Jr., R.E. Design of Purine Nucleoside Phosphorylase Inhibitors. Symposium: "Design of Antitumor Agents Based on Mechanism of Action," Fed. Proc. 45: 2773-2778 (1986). 113.Carson, W.M. and Bugg, C.E. An Algorithm for Ribbon Models of Proteins. J. Mol. Graphics 4: 121-122 (1986). 114.Bugg, C.E. Protein Crystal Growth in Space. Proc. Pharm. Tech. Con. '85, Cherry Hill, NJ, Sept. 10-12, pp. 22-26 (1985). 115.Montgomery, J.A., Thomas, H.J., Zell, A.L., Einspahr, H.M . and Bugg, C.E. Study on the Inhibition of Adenosine Deaminase. J. Med. Chem. 28: 1751-1753 (1985). 116.Bugg, C.E. The Future of Protein Crystal Growth. J. Cryst. Growth 76: 535-544 (1986). 117.DeLucas, L.J., Suddath, F.L., Snyder, R., Naumann, R., Broom, M.B., Pusey, M., Yost, V., Herren, B., Carter, D.C, Nelson, B., Meehan Jr., E.J., McPherson, A. and Bugg, C.E. Preliminary Investigations of Protein Crystal Growth Using the Space Shuttle. J. Cryst. Growth 76: 681-693 (1986). 118.DeLucas, L.J., Suddath, F.L., Snyder, R., Naumann, R., Broom, M.B., Pusey, M., Yost, V., Herren, B., Carter, D., Nelson, B., Meehan Jr., E.J., McPherson, A. and Bugg, C.E. Protein Crystal Growth in Microgravity. Space: Biomedicine and Biotechnology Proceedings, Ottawa, Ontario, May 29-30, 1987. 119.Vijay-Kumar, S., Bugg, C.E. and Cook, W.J. Three-Dimensional Structure of Ubiquitin at 1.8A Resolution. J. Mol. Biol. 194: 531-544 (1987). 120.Vijay-Kumar, S., Senadhi, S.E., Ealick, S.E., Nagabhushan, T.L., Trotta, P.P., Kosecki, R., Reichert, P. and Bugg, C.E. Crystallization and Preliminary X-ray Investigation of a Recombinant Form of Human Gamma-Interferon. J. Biol. Chem. 262: 4804-4805 (1987). 121.Vijay-Kumar, S., Bugg, C.E., Wilkinson, K.D., Vierstra, R.D., Hatfield, P.M. and Cook, W.J. Comparison of the Three-Dimensional Structures of Human, Yeast and Oat Ubiquitin. J. Biol. Chem. 262: 6396-6399 (1987). 122.DeLucas, L.J., Suddath, F.L., Snyder, R., Naumann, R., Broom, M.B., Pusey, M., Yost, V., Herren, B. and Bugg, C.E. Protein Crystal Growth Using the Space Shuttle. Polym. Prepr. 28: 383-384 (1987). 123.DeLucas, L.J., Greenhough, T.J., Rule, S.A., Myles, D.A., Babu, Y.S., Volanakis, J.E. and Bugg, C. E. Preliminary X-ray Study of Crystals of Human C-Reactive Protein. J. Mol. Biol. 196: 741-742 (1987). 124.Warren, T.K., Wells, J., Panchal, R.G., Stuthman, K.S., Garza, N.L., Van Tongeren, S.A., Long, L., Retterer, C.J., Eaton, B.P., Pegoraro, G., Honnold, S., Bantia, S., Kotian, P., Chen, X., Taubenheim, B.R., Welch, L.S., Minning, D.M., Babu, Y.S., Sheridan, W.P., and Bavari, S. Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX-4430, Nature 508:402-405 (2014). 125.Babu, Y.S., Bugg, C.E. and Cook, W.J. Crystal Structure of Calmodulin. In: Calcium Binding Proteins 1986: Proceedings of the Fifth International Symposium on Calcium Binding Proteins in Health and Disease (ed . N . Anthony et al.), Academic Press, NY, pp. 305-311 (1987). 126.Babu, Y.S., Bugg, C.E. and Cook, W.J. Three-Dimensional Structure of Calmodulin. In: Molecular Aspects of Cellular Recognition, Vol. 5 (Calmodulin), pp. 83-89 (1988). 127.Babu, Y.S., Bugg, C.E. and Cook, W.J. X-ray Diffraction Studies of Calmodulin. Methods Enzym. 139: 632-642 (1987). 128.DeLucas, L.J. and Bugg, C.E. New Directions in Protein Crystal Growth. Trends Biotechnol. 5: 188-193 (1987). 129.Babu, Y.S., Bugg, C.E . and Cook, W.J. Structure of Calmodulin Refined at 2.2A Resolution. J. Mol. Biol. 204: 191-204 (1988). 130.Bugg, C.E. Crystallization of Proteins and Other Biological Macromolecules. In: Crystal Growth in Science and Technology, Series B: Physics Vol. 210 (ed. H. Arend and J. Hulliger), Plenum Press (1989). 131.Krishna, N.R., Nettesheim, D.C., Klevit, R.E., Drobny, G., Watt, D.D. and Bugg, C.E. Proton Nuclear Magnetic Resonance Characterization of the Aromatic Residues in the Variant-3 Neurotoxin from Centruroides sculpturatus Ewing. Biochem. 28: 1556-1562 (1989) . 132.DeLucas, L.J., Smith, C.O., Smith, H.W., Vijay-Kumar, S., Senadhi, S.E., Ealick, S.E., Carter, D.C, Snyder, R.S., Weber, P.C, Salemme, F.R, Ohlendorf, D.H., Einspahr, H.M., Navia, M.A, McKeever, B.M., Nagabhushan, T.L., Nelson, G., McPherson, A., Koszelak, S., Taylor, G., Stammers, D., Powell, K., Darby, G. and Bugg, C.E. Protein Crystal Growth in Microgravity. Science 246: 651-654 (1989). 133.Bugg, C.E. Crystallization of Proteins and Other Biological Molecules. NATO ASI Ser. B210: 239- 251 (1989). 134.Rowland, R.S., Allen, F.H., Carson, W.M. and Bugg, C.E. Preferred Interaction Patterns from Crystallographic Databases. In: Proceedings of the Crystallographic and Modeling Methods in Molecular Design Symposium (ed . C.E. Bugg and S.E. Ealick), Springer-Verlag, NY, pp. 229-253 (1990). 135.Ealick, S.E., Babu, Y.S., Narayana, S.V.L., Cook, W.J. and Bugg, C.E. Design of Purine Nucleoside Phosphorylase Inhibitors Using X-ray Crystallography. In: Proceedings of the Crystallographic and Modeling Methods in Molecular Design Symposium (ed. C.E. Bugg and S.E. Ealick), Springer-Verlag, NY, pp. 43-55 (1990). 136.Rowland, R.S., Carson, W.M. and Bugg, C.E. Applications of Crystallographic Databases in Molecular Design. In: The Use of X-ray Crystallography in the Design of Anti-Viral Agents (ed. W.G. Laver and G.M. Air), Academic Press (1990). 137.Reichert, P., Ealick, S.E., Cook, W.J., Trotta, P., Nagabhushan, T.L. and Bugg, C.E. Crystallization and Preliminary X-ray Investigation of Recombinant Human Granulocyte-Macrophage Colony- Stimulating Factor. J. Biol. Chem. 265: 452-453 (1990). 138.Myles, D.A, Rule, S.A, DeLucas, L.J., Babu, Y.S., Xu, Y., Volanakis, J.E., Bugg, C.E., Bailey, S. and Greenhough, T.J. Rotation Function Studies of Human C-Reactive Protein. J. Mol. Biol. 216: 491-496 (1990). 139.Ealick, S.E., Rule, S.A , Carter, D.C, Greenhough, T.J., Babu, Y.S., Cook, W.J., Habash, J., Helliwell, J.R, Stoeckler, J.D., Parks Jr., R.E., Chen, S-F. and Bugg, C.E. Three-Dimensional Structure of Human Erythrocytic Purine Nucleoside Phosphorylase at 3.2A Resolution. J. Biol. Chem. 265: 1812- 1820 (1990). 140.Bugg, C.E. and DeLucas, L.J. Protein Crystal Growth in Space. In: Space Commerce '90 (ed . J.J. Egan), Gordon & Breach (1990). 141.Ealick, S.E., Babu, Y.S., Narayana, S.V.L., Cook, W.J. and Bugg, C.E. Rational Drug Design for the Treatment of AIDS Using X-ray Crystallography. In: Advances in Chemotherapy of AIDS (ed. R.B. Diasio and J.-P. Sommadossi), Pergamon Press (1990). 142.DeLucas, L.J. and Bugg, C.E. Protein Crystal Growth Procedure for the U.S. Space Shuttle. In: Methods: A Companion to Methods in Enzymology, Vol. I, No. I , pp. 105-109 (1990). 143.DeLucas, L.J., Smith, C.D., Carter, D.C, Snyder, R,S., McPherson, A , Koszelak, S. and Bugg, C.E. Microgravity Protein Crystal Growth: Results and Hardware Development. J. Cryst. Growth 109: 12-16 (1991). 144.DeLucas, L.J., Smith, C.D., Smith, H.W., Vijay-Kumar, S., Senadhi, S.E., Ealick, S.E., Carter, D.C, Snyder, R.S., Weber, P.C, Salemme, F.R, Ohlendorf, D.H., Einspahr, H.M., Clancy, L.L., Navia, M.A, McKeever, B.M., Nagabhushan, T.L., Nelson, G., McPherson, A, Koszelak, S., Taylor, G., Stammers, D., Powell, K., Darby, G. and Bugg, C.E. Protein Crystal Growth Results for Shuttle Flights STS-26 and STS-29. J. Cryst. Growth 110: 302-311 (1991). 145.DeLucas, L.J. and Bugg, C.E. Protein Crystal Growth in Space. In: Adv. Space Biol. & Med., Vol. I, pp. 249-278 (JAI Press) (1991). 146.Bugg, C.E. and DeLucas, L.J. Protein Crystal Growth in Microgravity. J. Clin. Phar. 31 : 985-987 (1991). 147.Bugg, C.E. and DeLucas, L.J. Microgravity Protein Crystal Growth. AIAA Journal (1991). 148.El-Kabbani, O., Sthanam, V.L.N., Babu, Y.S., Moore, K.M ., Flynn, T.G., Petrash, J.M., Westbrook, E.M., DeLucas, L.J. and Bugg, C.E. Purification, Crystallization and Preliminary Crystallographic Analysis of Porcine Aldose Reductase. J. Mol. Biol. 218: 695-698 (1991). 149.Cook, W.J., Ealick, S.E., Reichert, P., Hammond, G.S., Le, H.V., Nagabhushan, T.L., Trotta, P.P. and Bugg, C.E. Crystallization and Preliminary X-ray Investigation of Recombinant Human Interleukin 4. J. Mol. Biol. 218: 675-678 (1991). 150.Narayana, S.V.L., Kilpatrick, J.M., El-Kabbani, O., Babu, Y.S., Bugg, C.E., Volanakis, J.E. and DeLucas, L.J. Crystallization and Preliminary X-ray Investigation of Factor D of Human Complement. J. Mol. Biol. 219: 1-3 (1991). 151.Ealick, S.E., Babu, Y.S., Bugg, C.E., Erion, M.D., Guida, W.C, Montgomery, J.A and Secrist, J.A. Application of Crystallographic and Modeling Methods in the Design of Novel Purine Nucleoside Phosphorylase Inhibitors. Proc. Nat. Acad. Sci. USA 88: 11540-11544 (1991). 152.Carson, M. and Bugg, C.E. Analysis of the Protein Backbone. IEEE Computer Graphics & Applications, Vol. 12(1), pp. 11-13 (1992). 153.Ealick, S.E., Cook, W.J., Senadhi, V-K., Carson, M., Nagabhushan, T.L., Trotta, P.P. and Bugg, C.E. Three-Dimensional Structure of Recombinant Human Interferon-gamma. Science 251: 698-702 (1992). 154.DeLucas, L.J., Smith, C.D., Carter, D.C, Twigg, P., He, X-M., Snyder, R.S., Weber, P.C, Salemme, F.R, Ohlendorf, D.H., Einspahr, H.M., Clancy, L.L., McPherson, A., Koszelak, S., Vandonselaar, M.M., Prasad, L., Quail, J.W., Delbaere, L.T.J. and Bugg, C.E. Protein Crystal Growth Aboard the U.S. Space Shuttle Flights STS-31 and STS-32 . In: Adv. Space Res., Vol 123, pp. (1)393-(1)400 (1992). 155.Walter, M.R, Cook, W.J., Ealick, S.E., Nagabhushan, T.L., Trotta, P.P. and Bugg, C.E. Three-Dimensional Structure of Recombinant Human Granulocyte-Macrophage Colony-Stimulating Factor. J. Mol. Biol. 224: 1075-1085 (1992). 156.Walter, M.R, Cook, W.J., Zhao, B-G., Cameron, R.P., Reichert, P., Nagabhushan, T.L., Trotta, P.P. and Bugg, C.E. Crystal Structure of Recombinant Human Interleukin-4. J. Biol. Chem. 267: 20371-20376 (1992). 157.Zhao, B-G., Carson, W.M., Ealick, S.E. and Bugg, C.E. Structure of Scorpion Toxin Variant-3 at 1.2A Resolution. J. Mol. Biol. 227: 239-252 (1992). 158.Montgomery, J.A., Niwas, S., Rose, J.D., Secrist III, J.A , Babu, Y.S., Bugg, C.E., Erion, M.D., Guida, W.C and Ealick, S.E. Structure-Based Design of Inhibitors of Purine Nucleoside Phosphorylase. 1. 9-(arylmethyl) Derivatives of 9-Deazaguanine. J. Med. Chem. 26: 55-69 (1993). 159.DeLucas, L.J., Moore, K.M., Narayana, S.V.L., Bray, T.L., Rosenblum, W.M., Einspahr, H.M., Clancy, L.L., Rao, G.S.J., Harris, G.C., Munson, S.H., Finzel, B.C., Carter, D.C. and Bugg, C.E. Protein Crystal Growth Experiments and Results from the United States Microgravity Laboratory-1 Mission. J. Physics D: Applied Physics 26: B100-B103 (1993). 160.Ealick, S.E., Babu, Y.S., Bugg, C.E., Erion, M.D., Guida, W.G., Montgomery, J.A and Secrist, III, J.A Application of X-ray Crystallographic Methods in the Design of Purine Nucleoside Phosphorylase Inhibitors. Ann. N. Y. Acad. Sci. 685: 237-247 (1993). 161.Secrist, III, J.A, Niwas, S., Rose, J.D., Babu, Y.S., Bugg, C.E., Erion, M.D., Guida, W.C., Ealick, S.E. and Montgomery, J.A. Structure-Based Design of Inhibitors of Purine Nucleoside Phosphorylase. 2. 9-Alicyclic- and Heteroalicyclic Derivatives of 9-Deazaguanine. J. Med. Chem. 36: 1847-1854 (1993). 162.Erion, M.D., Niwas, S., Rose, J.D., Ananthan, S., Allen, M., Secrist III, J.A., Babu, Y.S., Bugg, C.E., Guida, W.C., Ealick, S.E. and Montgomery, J.A Structure-Based Design of Inhibitors of Purine Nucleoside Phosphorylase. 3.9-Arylmethyl Derivatives of 9-Deazaguanine Substituted on the Methyl Group. J. Med. Chem. 36: 3771 -3783 (1993). 163.Narayana, S.V.L., Carson, W.M., El-Kabbani, 0., Kilpatrick, J.M., Moore, D., Chen, X., Bugg, C.E., Volanakis, J.E. and DeLucas, L.J. Structure of Human Factor D, A Complement System Protein at 2.0A Resolution. J. Mol. Biol. 235: 695-708 (1994). 164.Bugg, C.E., Carson, W.M. and Montgomery, J.A. Drugs by Design. Sci. Amer. 269(6): 92-98 (1993). 165.DeLucas, L.J., Long, M.M., Moore, K.M., Rosenblum, W.M., Bray, T.L., Smith, C., Carson, M., Narayana, S.V.L., Carter, D.C., Clark, Jr., A.D., Nanni, R.G., Ding, J., Jacobo-Molina, A., Kamer, C., Hughes, S.H., Arnold, E., Einspahr, H.M., Clancy, L.L., Rao, G.S.J., Cook, P.F., Harris, B.C., Munson, S.H., Finzel, B.C., McPherson, A., Weber, P.C., Lewandowski, F.A., Nagabhushan, T.L., Trotta, P.P., Reichert, P., Navia, M.A., Wilson, K.P., Thomson, J.A., Richard, R.R., Bowersox, K.D., Meade, C.J., Baker, E.S., Bishop, S.P., Dunbar, B.J., Trinh, E., Prahl, J., Sacco Jr., A. and Bugg, C.E. Recent Results and New Hardware Developments for Protein Crystal Growth in Microgravity. J. Cryst. Growth 135: 183-195 (1994). 166.Guida, W.C., Elliott, RD., Thomas, H.J., Secrist III, J.A., Babu, Y.S., Bugg, C.E., Erion, M.D., Ealick, S.E. and Montgomery, J.A. Structure-Based Design of Inhibitors of Purine Nucleoside Phosphorylase. 4. A Study of Phosphate Mimics. J. Med. Chem. 37: 1109-1114 (1994). 167.Carson, M., Bugg, C.E., Narayana, S.V.L. and DeLucas, L.J. Comparison of Homology Models with the Experimental Structure of a Novel Serine Protease. Acta Cryst D50: 889-899 (1994). 168.Carson, M., Buckner, T.W., Yang, Z., Narayana, S.V.L. and Bugg, C.E. Error Detection in Crystallographic Models. Acta Cryst D50: 900-909 (1994). 169.Betzel, C., Gunther, N ., Poll, S., Moore, K, DeLucas, L.J., Bugg, C.E. and Weber, W. Crystallization of the EGF Receptor Ectodomain on US Space Mission STS-47. Microgravity Sci. Technol. VII/3: 242- 245 (1994). 170.Babu, Y.S., Ealick, S.E., Bugg, C.E., Erion, M.D., Guida, W.C., Montgomery, J.A. and Secrist, III, J.A. Structure-Based Design of Inhibitors of Purine Nucleoside Phosphorylase. Acta Cryst D51 :529-535 (1995). |